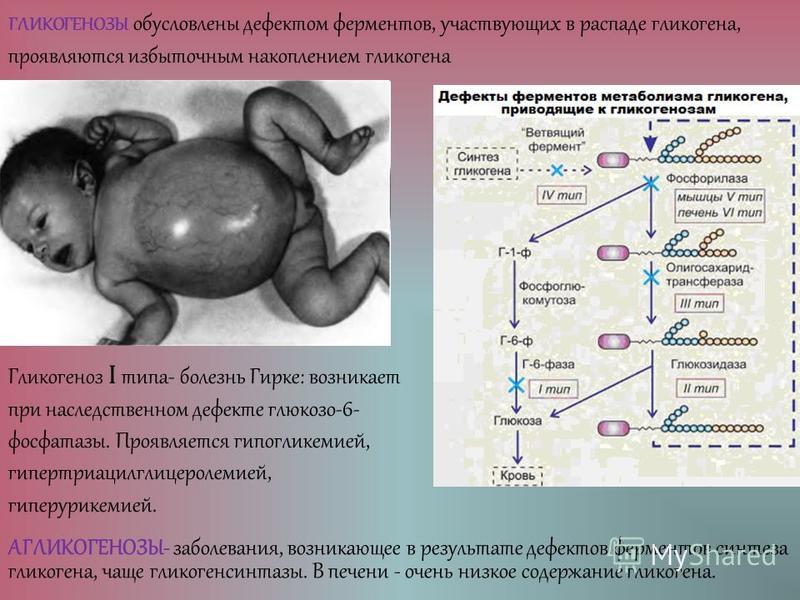
Гликогенозы у детей: Гликогенозы. Что такое Гликогенозы?
Гликогенозы. Что такое Гликогенозы?
ВАЖНО
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Гликогенозы – наследственные болезни, в основе которых лежит генетический дефект производства ферментов, принимающих участие в метаболизме углеводов. Характерный общий признак – чрезмерное отложение гликогена в миоцитах, гепатоцитах и других клетках организма. Гликогенозы проявляются симптомами гипогликемии, гепатомегалии, мышечной слабости, печеночной, сердечной, дыхательной и почечной недостаточности. Диагностика включает биохимический анализ крови, морфологическое исследование биопсийного материала мышц и печени, определение активности ферментов, молекулярно-генетические тесты. Лечение основано на лечебном питании, медикаментозной коррекции метаболических расстройств, в ряде случаев требуются операции.
МКБ-10
E74.0 Болезни накопления гликогена
- Причины гликогенозов
- Патогенез
- Классификация
- Симптомы гликогенозов
- Осложнения
- Диагностика
- Лечение гликогенозов
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Исследование гликогенозов ведется с 1910 года. В 1928-29 годах была описана симптоматика гликогеноза I типа – «болезни накопления гликогена». Лишь в 1952 году удалось выявить ферментный дефект и установить его связь с развитием симптомов. Патогенетические механизмы и способы лечения до сих пор остаются не до конца изученными. К настоящему времени выделено 12 типов гликогенозов, наиболее полно исследовано 9. Распространенность низкая, в среднем составляет 1 случай на 40-68 тысяч населения. Эпидемиологические показатели одинаковы среди представителей обоих полов, но при X-рецессивном наследовании мужчины болеют чаще.
Гликогенозы
Причины гликогенозов
Единственным фактором, провоцирующим развитие гликогеновых болезней, является генетический дефект, в результате которого возникает недостаточность определенного фермента, участвующего в обмене глюкозы. Все гликогенозы за исключением IX типа наследуются по аутосомно-рецессивному принципу. Это означает, что мутационный ген расположен на хромосоме, не сцепленной с полом, проявление заболевания возможно только при наследовании мутаций от каждого из родителей – при наличии двух рецессивных измененных генов в аллели. Если дефектным является один ген из пары, то другой – доминантный, нормальный – обеспечивает организм достаточным количеством фермента. Человек при этом становится носителем гликогеноза, но не болеет. В парах, где оба партнера – носители, вероятность рождения больного ребенка составляет 25%.
Патогенез
Патогенетическая основа всех гликогенозов – невозможность процесса синтеза и распада гликогена, его накопление в тканях. Гликоген является единственным резервным полисахаридом организма, своеобразным энергетическим «депо» – после приема пищи излишек глюкозы превращается в гликоген печени и мышц, затем постепенно расщепляется обратно до глюкозы. Благодаря этому механизму поддерживается стабильный уровень сахара в плазме крови, клетки и ткани организма непрерывно обеспечиваются энергией. При агликогенозе (0 тип) – отсутствует фермент гликогенсинтетаза, ответственная за производство гликогена. Пациенты страдают от тяжелой гипогликемии.
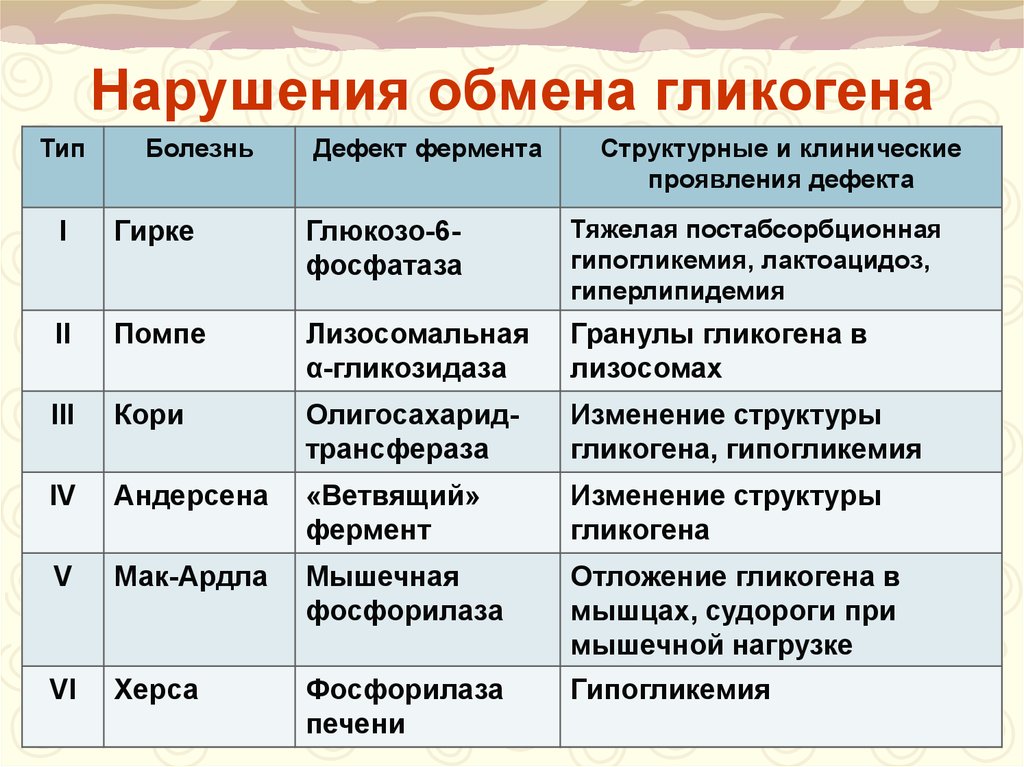
При гликогеновых болезнях типов 1-11 возникает генетически обусловленная недостаточность какого-либо фермента, катализирующего цепочку глюкоза-гликоген-глюкоза. 1 тип характеризуется дефектом глюкозо-6-фосфатазы и глюкозо-6-фосфаттранслоказы, 2 тип – альфа-1,4-глюкозидазы, 3 тип – амило-1,6-глюкозидазы, 4 тип – D-1,4-глюкано-α-глюкозилтрансферазы, 5 тип – гликогенфосфорилазы миоцитов, 6 тип – крахмалфосфорилазы гепатоцитов, 7 тип – фосфоглюкомутазы, 8 тип – фосфофруктомутазы, 9 тип – киназы фосфорилазы гепатоцитов. Из-за сниженной активности или полного отсутствия фермента гликоген накапливается в мышцах, печени, редко – в других тканях. Изменяется структура и функциональность органов, развиваются различные формы органной недостаточности.
1 тип характеризуется дефектом глюкозо-6-фосфатазы и глюкозо-6-фосфаттранслоказы, 2 тип – альфа-1,4-глюкозидазы, 3 тип – амило-1,6-глюкозидазы, 4 тип – D-1,4-глюкано-α-глюкозилтрансферазы, 5 тип – гликогенфосфорилазы миоцитов, 6 тип – крахмалфосфорилазы гепатоцитов, 7 тип – фосфоглюкомутазы, 8 тип – фосфофруктомутазы, 9 тип – киназы фосфорилазы гепатоцитов. Из-за сниженной активности или полного отсутствия фермента гликоген накапливается в мышцах, печени, редко – в других тканях. Изменяется структура и функциональность органов, развиваются различные формы органной недостаточности.
Классификация
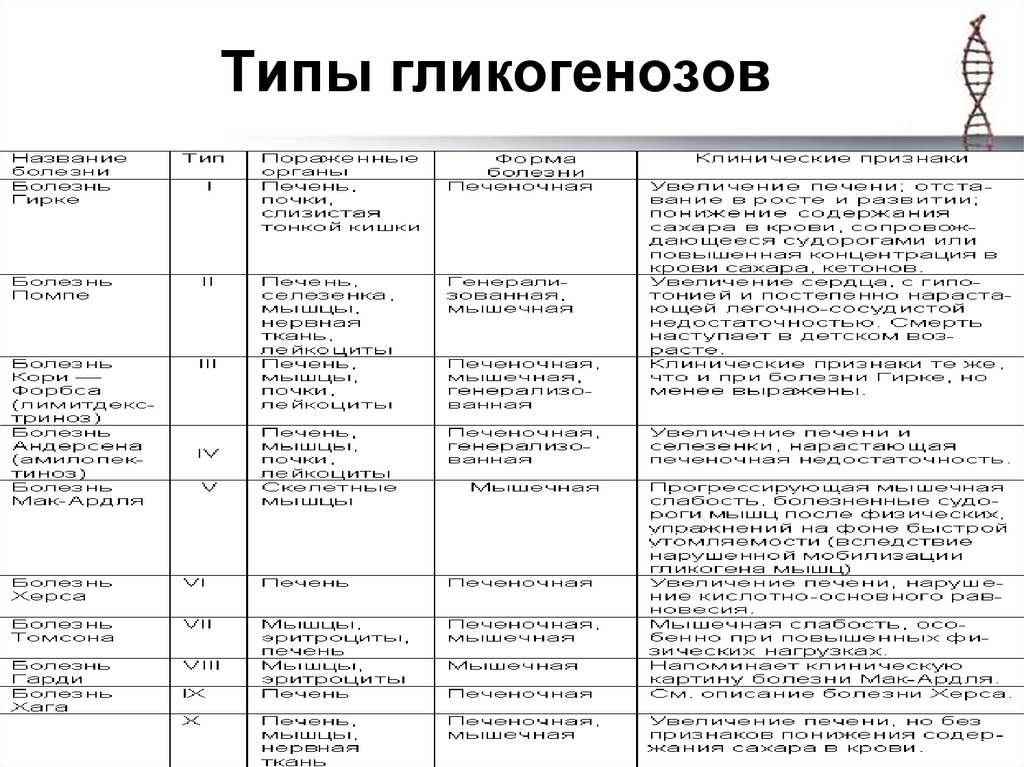
С учетом ферментативного дефекта и особенностей клинических проявлений выделяют 12 вариантов гликогенозов, от 0 до XI. Кроме того, описаны случаи комбинированных типов, когда определяется дефицит двух ферментов, а также случаи неидентифицируемых типов, при которых выделить ферментный дефект не удается. Согласно ведущему патогенетическому механизму гликогеновые болезни подразделяются на три больших группы:
- Печеночные.
 Включают гликогенозы всех типов, кроме II, V и VII. Гликоген откладывается преимущественно в гепатоцитах. Характерна гепатомегалия, гипогликемия через 2 часа после поступления углеводов. При I типе заболевания также поражаются почки, при III и IV типах развиваются миопатии.
Включают гликогенозы всех типов, кроме II, V и VII. Гликоген откладывается преимущественно в гепатоцитах. Характерна гепатомегалия, гипогликемия через 2 часа после поступления углеводов. При I типе заболевания также поражаются почки, при III и IV типах развиваются миопатии. - Мышечные. В данную группу входят болезни типов VII и V. Изменена ферментативная активность в мышечной ткани, нарушено энергообеспечение мышц. Типичные симптомы – миалгии, судороги.
- Смешанные. Гликогеноз II типа отличается тем, что в патологический процесс вовлекаются все гликогенсодержащие ткани. Гликоген скапливается в лизосомах и цитоплазме клеток. Страдают многие органы, возрастает риск смерти по причине сердечной или дыхательной недостаточности.
 Включают гликогенозы всех типов, кроме II, V и VII. Гликоген откладывается преимущественно в гепатоцитах. Характерна гепатомегалия, гипогликемия через 2 часа после поступления углеводов. При I типе заболевания также поражаются почки, при III и IV типах развиваются миопатии.
Включают гликогенозы всех типов, кроме II, V и VII. Гликоген откладывается преимущественно в гепатоцитах. Характерна гепатомегалия, гипогликемия через 2 часа после поступления углеводов. При I типе заболевания также поражаются почки, при III и IV типах развиваются миопатии.Симптомы гликогенозов
Агликогеноз развивается в периоде новорожденности либо раннего детства. Низкое содержание гликогена в печени проявляется резко выраженной гипогликемией натощак. Наблюдается заторможенность, глубокий сон, потеря сознания, бледность кожи, тошнота, рвота, судороги ночью и в утренние часы.
Клинические признаки болезни Помпе (II тип) определяются в течение нескольких недель после рождения. Дети вялые, малоподвижные, с ослабленным сосательным рефлексом, сниженным аппетитом. Гепатомегалия изменяет пропорции тела – живот увеличивается, руки и ноги остаются тонкими. Поражается сердце, легкие, нервная система. Высок риск сердечной и легочной недостаточности. У пациентов с болезнью Форбса (III тип) симптомы слабой и умеренной выраженности. На первый план выходит гипогликемия постабсорбционного периода, гепатомегалия, накопление подкожного жира в области туловища. Ведущие симптомы болезни Андерсена (IV тип)– мышечная слабость, плохая переносимость физической нагрузки, судороги.
Болезнь Томсона представлена гепатомегалией, нистагмом, атаксией, прогрессирующими неврологическими нарушениями с мышечной гипертонией, децеребрацией. Типичные проявления болезни Мак-Ардля (V тип) – боли, спазматические сокращения, чрезмерная утомляемость и слабость мышц даже после незначительной нагрузки. Иногда тонические судороги переходят в генерализованные, что сопровождается общей скованностью. Проявления болезни Герса (VI тип) менее выраженные, пациенты способны переносить легкие и умеренные физические нагрузки, не испытывая дискомфорта. Дополнительно обнаруживаются признаки поражения печени – угнетение аппетита, рвота, тошнота, боли в правом боку.
Течение болезни Таруи (VII тип) включает непереносимость физической нагрузки, сопровождающуюся тошнотой и рвотой, болезненными спазмами мышц. Поступление глюкозы не повышает способность совершать физические действия. После употребления пищи симптомы обостряются. Наиболее мягкое течение свойственно болезни Хага (IX тип).
Осложнения
При разновидностях гликогенозов, сопровождающихся гипогликемией, существует риск развития гипогликемической комы. Как правило, выраженное снижение уровня глюкозы в крови происходит при пропуске приемов пищи, особенно после ночного сна (пропуск завтрака). Пациенты испытывают головокружение и судороги, теряют сознание. Тяжелые формы мышечных гликогенозов при продолжительном течении и отсутствии терапии приводят к дистрофии скелетных мышц, сердечной недостаточности. Осложнением некоторых печеночных гликогенозов является цирроз печени.
Диагностика
При подозрении на гликогеноз ребенку рекомендуется консультация врача-генетика, педиатра, гастроэнтеролога, гепатолога. В первую очередь специалист собирает анамнез, проводит клинический опрос и осмотр. Поскольку заболевание передается аутосомно-рецессивным способом, семейные случаи выявляют редко. Распространены жалобы на слабость, апатичность ребенка, бледность и желтушность кожи, отказ от еды или повышенный аппетит, трудности пробуждения утром, тремор, судороги. При осмотре врач отмечает увеличение размера печени, выпирание живота, задержку роста, мышечную гипотрофию, специфическое отложение подкожной жировой клетчатки, ксантомы. Лабораторные и инструментальные методы позволяют подтвердить диагноз гликогеноза, исключить врожденный сифилис, токсоплазмоз, цитомегалию, патологии печени, болезнь Гоше, миотонию, прогрессирующую мышечную дистрофию, амиотрофии. К обязательным методам исследований относят:
- Биохимическое исследование крови. По результатам анализа обнаруживается гипогликемия с уровнем глюкозы натощак 0,6-3 ммоль/л, лактатацидоз с концентрацией молочной кислоты 3-10 ммоль/л (кроме гликогеноза 4 типа). Дополнительно выявляется увеличение показателей триглицеридов, общего холестерина, ЛПНП, ЛПОНП, мочевой кислоты, печеночных ферментов.
- Исследование биоптата печени, мышц. При изучении ткани печени наиболее распространенными характеристиками являются повышенное количество гликогена и его глыбчатое распределение в цитоплазме гепатоцитов, иногда – в вакуолизированных ядрах. Определяется выраженная белковая и/или крупно- и мелкокапельная жировая дистрофия гепатоцитов, их некроз, ограниченные очаги фиброза в местах гибели клеток. Возможны признаки цирроза. При мышечных типах болезней исследуется мышечный биоптат, в котором просматриваются субсарколеммальные накопления структурно нормального гликогена.
- Исследование ферментов. Активность ферментов изучается в культуре кожных фибробластов, биоптате мышечной и печеночной ткани, лейкоцитах. При гликогенозах с хроническим медленно прогрессирующим течением снижение функциональности фермента легкое или умеренное. При тяжелом течении фермент отсутствует либо его активность минимальна.
- УЗИ брюшной полости. Отмечается выраженное увеличение печени, особенно левой ее доли. Характерна гиперэхогенность и структурная диффузная неоднородность паренхимы (множественные мелкие гиперэхогенные эхосигналы, распределенные равномерно). В дистальных отделах паренхимы прохождение ультразвука ослаблено. Возможно обнаружение структурно разнообразных печеночных аденом, увеличение размеров почек, селезенки и поджелудочной железы.
 По результатам анализа обнаруживается гипогликемия с уровнем глюкозы натощак 0,6-3 ммоль/л, лактатацидоз с концентрацией молочной кислоты 3-10 ммоль/л (кроме гликогеноза 4 типа). Дополнительно выявляется увеличение показателей триглицеридов, общего холестерина, ЛПНП, ЛПОНП, мочевой кислоты, печеночных ферментов.
По результатам анализа обнаруживается гипогликемия с уровнем глюкозы натощак 0,6-3 ммоль/л, лактатацидоз с концентрацией молочной кислоты 3-10 ммоль/л (кроме гликогеноза 4 типа). Дополнительно выявляется увеличение показателей триглицеридов, общего холестерина, ЛПНП, ЛПОНП, мочевой кислоты, печеночных ферментов. При гликогенозах с хроническим медленно прогрессирующим течением снижение функциональности фермента легкое или умеренное. При тяжелом течении фермент отсутствует либо его активность минимальна.
При гликогенозах с хроническим медленно прогрессирующим течением снижение функциональности фермента легкое или умеренное. При тяжелом течении фермент отсутствует либо его активность минимальна.Комплекс диагностических исследований подбирается индивидуально в зависимости от возраста пациента и предполагаемого типа гликогеновой болезни. Может потребоваться молекулярно-генетическая диагностика (секвенирование генов с целью выявления мутации), электромиография, ЭХО-КГ, ОАК, коагулограмма.
Лечение гликогенозов
Специфические методы терапии не разработаны.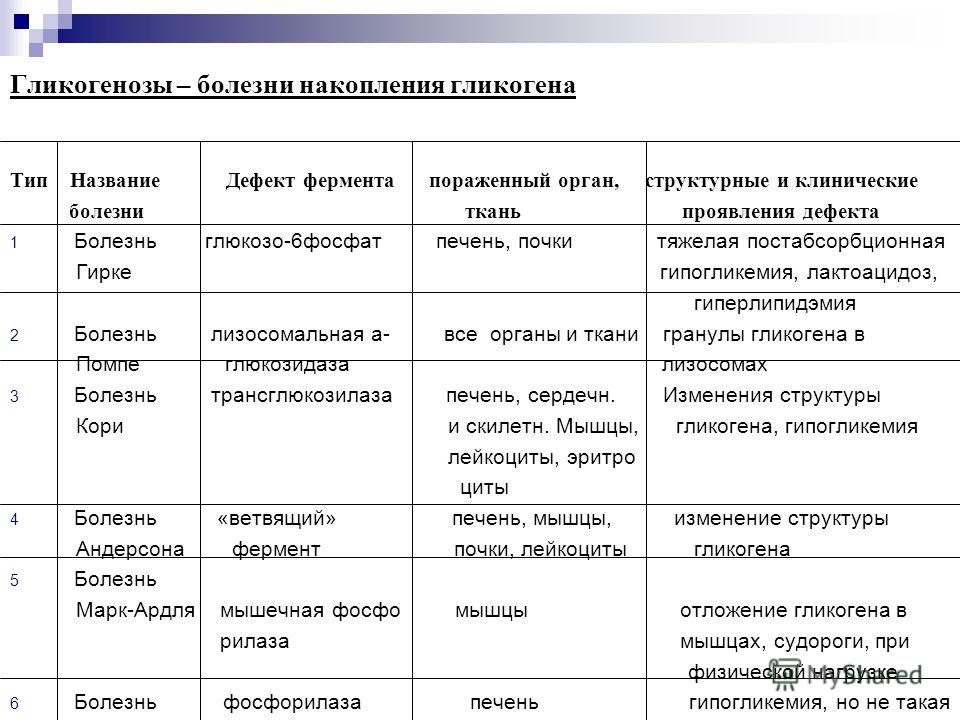 Патогенетическое лечение проводится консервативно, направлено на устранение гипогликемии, метаболического ацидоза, кетоза, гиперлипидемии, коррекцию дисфункции гепатобилиарного комплекса и желудочно-кишечного тракта. При развитии осложнений (серьезном поражении внутренних органов) выполняются хирургические операции. Медицинская помощь пациентам включает следующие направления:
Патогенетическое лечение проводится консервативно, направлено на устранение гипогликемии, метаболического ацидоза, кетоза, гиперлипидемии, коррекцию дисфункции гепатобилиарного комплекса и желудочно-кишечного тракта. При развитии осложнений (серьезном поражении внутренних органов) выполняются хирургические операции. Медицинская помощь пациентам включает следующие направления:
- Диетотерапию. Для минимизации метаболических нарушений составляется индивидуальный план питания. Больным рекомендуется снизить количество жиров, сахарозы, фруктозы и галактозы для уменьшения гиперлипидемии и ацидоза. При первом типе гликогеноза назначается диета с увеличенным потреблением углеводов. В частности, показано употребление сырого кукурузного крахмала с медленной усвояемостью, позволяющей предупредить гипогликемию. При типах 3, 4 и 9 вводится рацион с преобладанием животного белка и дробным питанием.
- Лекарственную коррекцию симптомов. В рамках комплексного лечения применяется кокарбоксилаза для увеличения производства ацетилкофермента А, кортикостероидов и глюкагона для стимуляции глюконеогенеза. Дефицит карнитина компенсируется левокарнитином. При вторичных тубулопатиях, печеночных и билиарных дисфункциях используются желчегонные препараты, гепатопротекторы, липотропные вещества. При признаках ацидоза показаны щелочные растворы внутривенно. При почечной дисфункции, протеинурии – ингибиторы АПФ. При гиперурикемии – урикодепрессоры. При нейтропении – гранулоцитарный колониестимулирующий фактор.
- Хирургическое лечение. Пациентам с тяжелыми фатальными поражениями печени может потребоваться ортотопическая трансплантация органа. Показанием к операции является цирроз с осложнениями, часто развивающийся при третьем и четвертом типе патологии. В отдельных случаях хирургическое вмешательство целесообразно при аденомах печени с высоким риском трансформации в злокачественную опухоль. Трансплантация почек иногда выполняется больным с хронической почечной недостаточностью.
 Дефицит карнитина компенсируется левокарнитином. При вторичных тубулопатиях, печеночных и билиарных дисфункциях используются желчегонные препараты, гепатопротекторы, липотропные вещества. При признаках ацидоза показаны щелочные растворы внутривенно. При почечной дисфункции, протеинурии – ингибиторы АПФ. При гиперурикемии – урикодепрессоры. При нейтропении – гранулоцитарный колониестимулирующий фактор.
Дефицит карнитина компенсируется левокарнитином. При вторичных тубулопатиях, печеночных и билиарных дисфункциях используются желчегонные препараты, гепатопротекторы, липотропные вещества. При признаках ацидоза показаны щелочные растворы внутривенно. При почечной дисфункции, протеинурии – ингибиторы АПФ. При гиперурикемии – урикодепрессоры. При нейтропении – гранулоцитарный колониестимулирующий фактор.Прогноз и профилактика
Эффективность терапии, вероятность осложнений и летального исхода зависят от типа патологии. Одни гликогенозы незначительно ухудшают качество жизни больных, компенсируются по мере взросления, другие – не поддаются лечению и неизбежно завершаются смертью. Для снижения риска рождения ребенка с гликогенозом супружеским парам из группы риска – имеющим семейный отягощенный анамнез, детей с подтвержденным диагнозом – требуется медико-генетическое консультирование, пренатальная диагностика.
Одни гликогенозы незначительно ухудшают качество жизни больных, компенсируются по мере взросления, другие – не поддаются лечению и неизбежно завершаются смертью. Для снижения риска рождения ребенка с гликогенозом супружеским парам из группы риска – имеющим семейный отягощенный анамнез, детей с подтвержденным диагнозом – требуется медико-генетическое консультирование, пренатальная диагностика.
Вы можете поделиться своей историей болезни, что Вам помогло при лечении гликогенозов.
Источники
- Педиатрия. Национальное руководство в 2 томах. Том 2./ под ред. Баранова А.А. — 2009.
- Гликогеновая болезнь у детей. Клинические рекомендации/ Союз педиатров России – 2016.
- Заболевания печени и желчных путей/ Шерлок Ш. Дули Дж. — 1999.
- Настоящая статья подготовлена по материалам сайта: https://www.krasotaimedicina.ru/
ВАЖНО
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Ведение детей с гликогеновой болезнью (нозологические формы с поражением печени). Современные клинические рекомендации | Баранов
1. Cori GT. Glycogen structure and enzyme dificiencies in Glycogen Storage Disease. Harvey Lect. 1952–1953;48:145–171.
2. Сурков А.Н. Гликогеновая болезнь у детей: новые аспекты патогенеза, современные подходы к диагностике, оптимизация ведения пациентов: автореф. дис. … докт. мед. наук. М.; 2019.
3. Ozen H. Glycogen storage diseases: new perspectives. World J Gastro enterol. 2007;13(18):2541–2553. doi: 10.3748/wjg.v13.i18.2541.
4. Lei KJ, Chen YT, Chen H, et al. Genetic basis of glycogen storage disease type 1a: prevalent mutations at the glucose-6-phosphatase locus. Am J Hum Genet. 1995;57(4):766–771.
5. Kishnani PS, Austin SL, Arn P, et al. Glycogen storage disease type III diagnosis and management guidelines. Genet Med. 2010;12(7):446–463. doi: 10.1097/GIM.0b013e3181e655b6.
Glycogen storage disease type III diagnosis and management guidelines. Genet Med. 2010;12(7):446–463. doi: 10.1097/GIM.0b013e3181e655b6.
6. Lerner A, Iancu TC, Bashan N, et al. A new variant of glycogen storage disease. Type IXc. Am J Dis Child. 1982;136(5):406–410. doi: 10.1001/archpedi.1982.03970410024004.
7. Hendrickx J, Willems PJ. Genetic deficiencies of the glycogen phosphorylase system. Hum Genet. 1996;97(5):551–556. doi: 10.1007/BF02281858.
8. Ellingwood SS, Cheng A. Biochemical and Clinical Aspects of Glycogen Storage Diseases. J Endocrinol. 2018;238(3):R131–R141. doi: 10.1530/JOE-18-0120.
9. Chen YT. Glycogen storage diseases. In: The Metabolic Bases of Inherited Disease. 8th ed. Scriver CR., Beaudet AL, Sly WS, Valle D, eds. NY: McGraw-Hill: 2000. pp. 1521–1551.
10. Зайчик А.Ш., Чурилов Л.П. Основы общей патологии: учебное пособие для студентов медицинских ВУЗов. Ч. 2. Основы патохимии. СПб.: ЭЛБИ; 2000. 688 с.
11. Brody LC, Abel KJ, Castilla LH, et al. Construction of a transcription map surrounding the BRCA1 locus of human chromosome 17. Genomics. 1995;25(1):238–247. doi: 10.1016/0888-7543(95)80131-5.
Construction of a transcription map surrounding the BRCA1 locus of human chromosome 17. Genomics. 1995;25(1):238–247. doi: 10.1016/0888-7543(95)80131-5.
12. Цыгин А.Н. Сочетанные заболевания печени и почек у детей // Клиническая нефрология. — 2009. — № 3. — С. 47–51.
13. Chen YT. Type I glycogen storage disease: kidney involvement, pathogenesis and its treatment. Pediatr Nephrol. 1991;5(1):71–76. doi: 10.1007/BF00852851.
14. Ueno N, Tomita M, Ariga T, et al. Impaired monocyte function in glycogen storage disease type Ib. Eur J Pediatr. 1986;145(4): 312–314. doi: 10.1007/BF00439409.
15. Leuzzi R, Banhegyi G, Kardon T, et al. Inhibition of microsomal glucose-6-phosphate transport in human neutrophils results in apoptosis: a potential explanation for neutrophil dysfunction in glycogen storage disease type 1b. Blood. 2003;101(6):2381–2387. doi: 10.1182/blood-2002-08-2576.
16. Shen J, Bao Y, Liu H-M, et al. Mutations in exon 3 of the glycogen debranching enzyme gene are associated with glycogen storage disease type III that is differentially expressed in liver and muscle. J Clin Invest. 1996;98(2):352–357. doi: 10.1172/JCI118799.
J Clin Invest. 1996;98(2):352–357. doi: 10.1172/JCI118799.
17. Li XH, Gong QM, Ling Y, et al. Inherent lipid metabolic dysfunction in glycogen storage disease IIIa. Biochem Biophys Res Commun. 2014;455(1–2):90–97. doi: 10.1016/j.bbrc.2014.10.096.
18. Moses SW, Parvari R. The variable presentations of glycogen storage disease type IV: a review of clinical, enzymatic and molecular studies. Curr Mol Med. 2002;2(2):177–188. doi: 10.2174/1566524024605815.
19. Burwinkel B, Bakker HD, Herschkovitz E, et al. Mutations in the liver glycogen phosphorylase gene (PYGL) underlying glycogenosis type VI (Hers disease). Am J Hum Genet. 1998;62(4):785–791. doi: 10.1086/301790.
20. Chang S, Rosenberg MJ, Morton H, et al. Identification of a mutation in liver glycogen phosphorylase in glycogen storage disease type VI. Hum Molec Genet. 1998;7(5):865–870. doi: 10.1093/hmg/7.5.865.
21. Hug G, Schubert WK, Chuck G. Deficient activity of dephoshophosphorylase kinase and accumulation of glycogene in the liver. J Clin Invest. 1969;48(4):704–715. doi: 10.1172/JCI106028.
J Clin Invest. 1969;48(4):704–715. doi: 10.1172/JCI106028.
22. Краснопольская К.Д. Наследственные болезни обмена веществ: справочное пособие для врачей. М.: РОО «Центр социальной адаптации и реабилитации детей «Фохат»; 2005. 364 с.
23. Розенфельд Е.Л., Попова И.А. Врожденные нарушения обмена гликогена. М.; 1989. 239 с.
24. Humbert M., Labrune P., Simonneau G. Severe pulmonary arterial hypertension in type 1 glycogen storage disease. Eur J Pediatr. 2002;161(Suppl 1):S93–S96. doi: 10.1007/s00431-002-1012-y.
25. Лобзин В.С., Сайкова Л.А., Шиман А.Г. Нервно-мышечные болезни. СПб.; Гиппократ. 1998. 224 с.
26. Visser G, Rake JP, Labrune P, et al. Consensus guidelines for management of glycogen storage disease type 1b — European Study on Glycogen Storage Disease Type 1. Eur J Pediatr. 2002;161 (Suppl 1):S120–S123. doi: 10.1007/s00431-002-1017-6.
27. Visser G, Rake JP, Fernandes J, et al. Neutropenia, neutrophil dysfunction, and infl ammatory bowel disease in glycogen storage disease type Ib: results of the European Study on Glycogen Storage Disease type I. J Pediatr. 2000;137(2):187–191. doi: 10.1067/mpd.2000.105232.
J Pediatr. 2000;137(2):187–191. doi: 10.1067/mpd.2000.105232.
28. Kure S, Hou DC, Suzuki Y, et al. Glycogen storage disease type Ib without neutropenia. J Pediatr. 2000;137(2):253–256. doi: 10.1067/mpd.2000.107472.
29. Melis D, Fulceri R, Parenti G, et al. Genotype/phenotype correlation in glycogen storage disease type 1b: a multicentre study and review of the literature. Eur J Pediatr. 2005;164(8):501–508. doi: 10.1007/s00431-005-1657-4.
30. Shen JJ, Chen YT. Molecular characterization of glycogen storage disease type III. Curr Mol Med. 2002;2(2):167–175. doi: 10.2174/1566524024605752.
31. L’Hermine-Coulomb A, Beuzen F, Bouvier R, et al. Fetal type IV glycogen storage disease: clinical, enzymatic, and genetic data of a pure muscular form with variable and early antenatal manifestations in the same family. Am J Med Genet. 2005;139A(2):118–122. doi: 10.1002/ajmg.a.30945.
32. Szymanska E, Szymanska S, Truszkowska G, et al. Variable clinical presentation of glycogen storage disease type IV: from severe hepatosplenomegaly to cardiac insufficiency.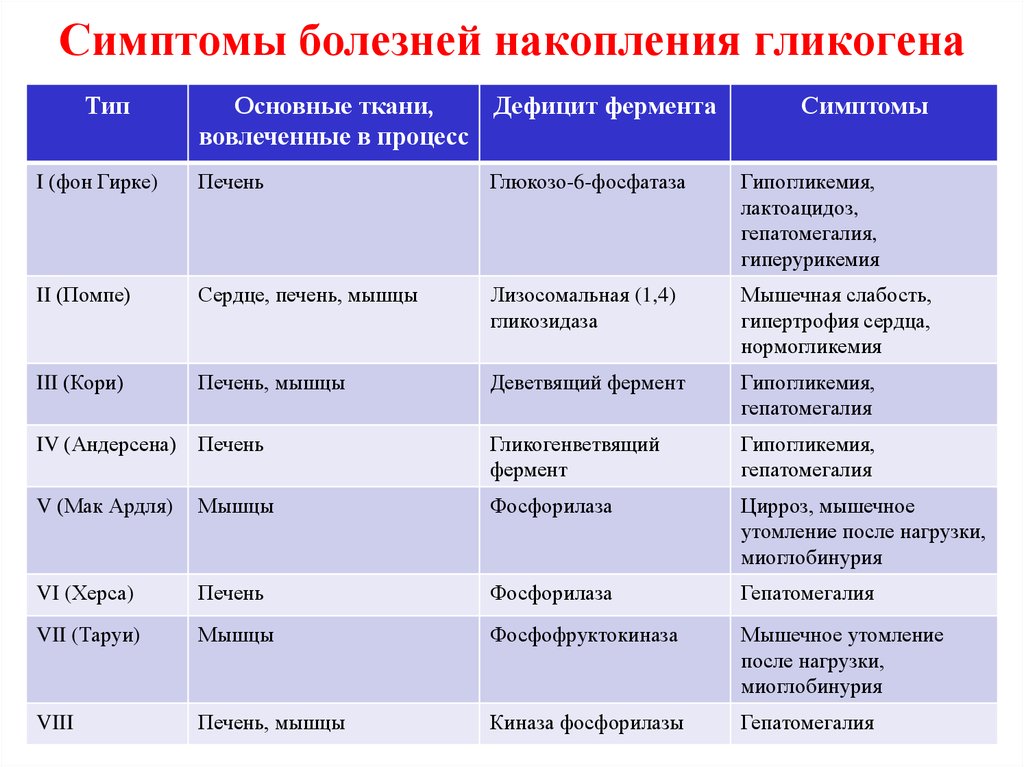 Some discrepancies in genetic and biochemical abnormalities. Arch Med Sci. 2018; 14(1):237–247. doi: 10.5114/aoms.2018.72246.
Some discrepancies in genetic and biochemical abnormalities. Arch Med Sci. 2018; 14(1):237–247. doi: 10.5114/aoms.2018.72246.
33. Malfatti E, Barnerias C, Hedberg-Oldfors C, et al. A novel neuromuscular form of glycogen storage disease type IV with arthrogryposis, spinal stiffness and rare polyglucosan bodies in muscle. Neuromuscul Disord. 2016;26(10):681–687. doi: 10.1016/j.nmd.2016.07.005.
34. Roscher A, Patel J, Hewson S, et al. The natural history of glycogen storage disease types VI and IX: Long-term outcome from the largest metabolic center in Canada. Mol Genet Metab. 2014; 113(3):171–176. doi: 10.1016/j.ymgme.2014.09.005.
35. Hodax JK, Uysal S, Quintos JB, Phornphutkul C. Glycogen storage disease type IX and growth hormone deficiency presenting as severe ketotic hypoglycemia. J Pediatr Endocrinol Metab. 2017;30(2): 247–251. doi: 10.1515/jpem-2016-0342.
36. Баранов А.А., Намазова-Баранова Л.С., Сурков А.Н. и др. Гликогеновая болезнь у детей: учебное пособие. М.: ПедиатрЪ; 2012. 128 с.
М.: ПедиатрЪ; 2012. 128 с.
37. Rake JP, Visser G, Labrune P, et al. Guidelines for management of glycogen storage disease type I — European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr. 2002;161(Suppl 1): S112–S119. doi: 10.1007/s00431-002-1016-7.
38. Laumonier H, Bioulac-Sage P, Laurent C, et al. Hepatocellular adenomas: magnetic resonance imaging features as a function of molecular pathological classification. Hepatology. 2008;48(3): 808–818. doi: 10.1002/hep.22417.
39. Дворяковская Г.М., Уварова Е.В., Дворяковский И.В. и др. Роль ультразвуковой диагностики при обследовании детей с печеночной формой гликогенозов // Ультразвуковая и функциональная диагностика. — 2002. — № 4. — С. 53–59.
40. Попович Ю.Г., Чибисов И.В., Потапова-Виноградова И.Н. и др. Клинико-биохимические и морфологические особенности печеночной формы гликогенозов у детей // Педиатрия. — 1988. — № 1. — С. 35–39.
41. Pathology of the liver. 4th ed. MacSween RNM, Burt AD, Portmann BC, eds.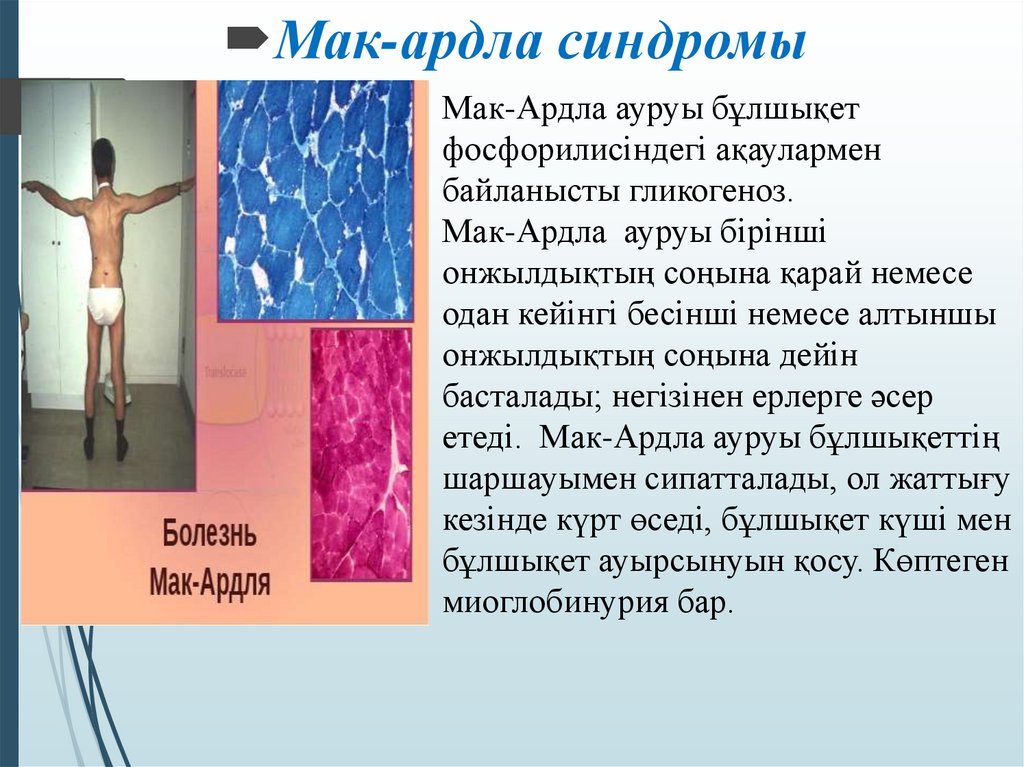 London: Churchill Livingstone; 2001. 982 p.
London: Churchill Livingstone; 2001. 982 p.
42. Клиническая диетология детского возраста: руководство для врача. Под ред. Т.Э. Боровик, К.С.Ладодо. М.: ООО «Медицинское информационное агентство»; 2008. 608 с.
43. Heller S, Worona L, Consuelo A. Nutritional therapy for glycogen storage diseases. J Pediatr Gastroenterol Nutr. 2008;47 (Suppl1): S15–S21. doi: 10.1097/MPG.0b013e3181818ea5.
44. Steunenberg TAH, Peeks F, Hoogeveen IJ, et. al. Safety issues associated with dietary management in patients with hepatic glycogen storage disease. Mol Genet Metab. 2018;125(1–2):79–85. doi: 10.1016/j.ymgme.2018.07.004.
45. Уварова Е.В. Течение гликогеновой болезни печени у детей в условиях комплексной терапии: автореф. дис. … канд. мед. наук. М., 2005. 28 с.
46. Correia CE, Bhattacharya K, Lee PJ, et al. Use of modified cornstarch therapy to extend fasting in glycogen storage disease types Ia and Ib. Am J Clin Nutr. 2008;88(5):1272–1276. doi: 10.3945/ajcn.2008.26352.
47. Visser G, Rake J, Labrune P, et al. Granulocyte colonystimulating factor in glycogen storage disease type 1b. Results of the European Study on Glycogen Storage Disease Type 1. Eur J Pediatr. 2002;161 (Suppl 1):S83–S87. doi: 10.1007/s00431-002-1010-0.
Visser G, Rake J, Labrune P, et al. Granulocyte colonystimulating factor in glycogen storage disease type 1b. Results of the European Study on Glycogen Storage Disease Type 1. Eur J Pediatr. 2002;161 (Suppl 1):S83–S87. doi: 10.1007/s00431-002-1010-0.
48. Melis D, Parenti G, Della Casa R, et al. Crohn’s-like ileo-colitis in patients affected by glycogen storage disease Ib: two years’ follow-up of patients with a wide spectrum of gastrointestinal signs. Acta Paediatr. 2003;92(12):1415–1421. doi: 10.1080/08035250310007033.
49. Готье С.В., Цирульникова О.М., Мнацаканян Д.С. и др. Трансплантация печени у детей с болезнями накопления гликогена: оценка риска и необходимость ее проведения // Вестник трансплантологии и искусственных органов. — 2013. — Т. XV. — № 1. — С. 67–74.
50. Филин А.В., Семенков А.В., Коротеева Н.А. и др. Родственная пересадка фрагментов печени при гликогенозах I типа: первый российский опыт // Трансплантология. — 2011. — № 2–3. — С. 24–28.
51. Boers SJ, Visser G, Smit PG, Fuchs SA.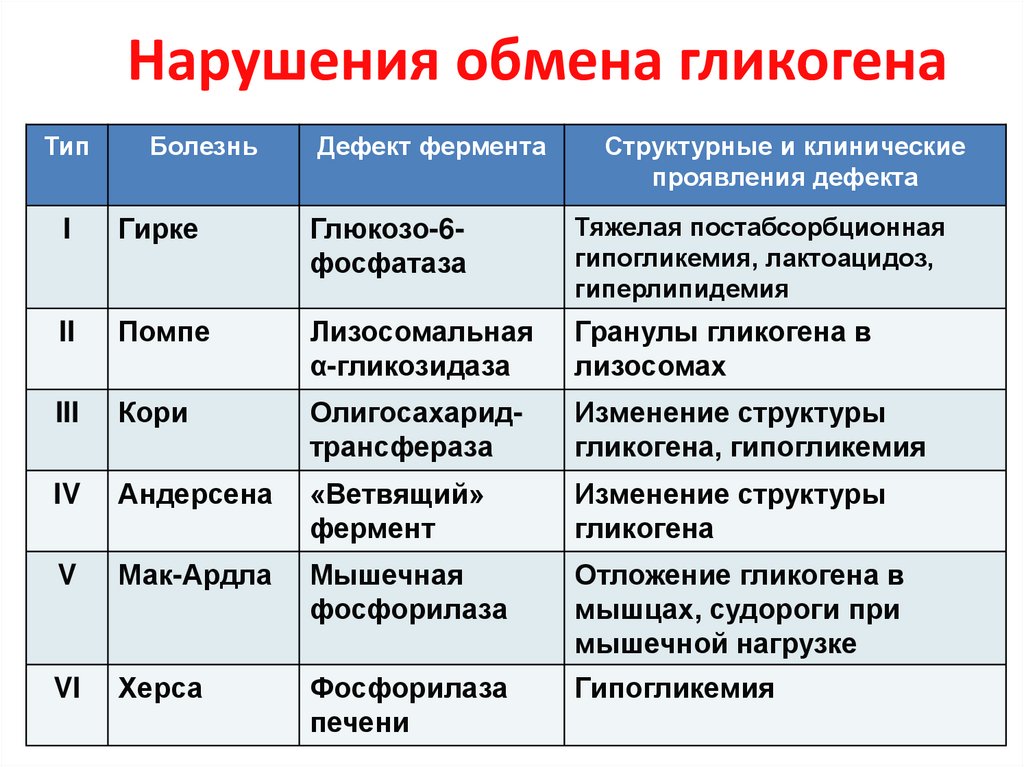 Liver transplantation in glycogen storage disease type I. Orphanet J Rare Dis. 2014;9:47. doi: 10.1186/1750-1172-9-47.
Liver transplantation in glycogen storage disease type I. Orphanet J Rare Dis. 2014;9:47. doi: 10.1186/1750-1172-9-47.
52. Oshita A, Itamoto T, Amano H, et al. Perioperative management of benign hepatic tumors in patients with glycogen storage disease type Ia. J Hepatobiliary Pancreat Surg. 2008;15(2):200–203. doi: 10.1007/s00534-007-1244-3.
Болезнь накопления гликогена | Boston Children’s Hospital
Болезнь накопления гликогена (GSD) — это название группы заболеваний, которые нарушают способность организма вырабатывать гликоген или преобразовывать гликоген в глюкозу. В зависимости от типа GSD у ребенка гликоген может накапливаться в печени, мышцах или и там, и там. GSD также может поражать клетки крови, сердце, почки и другие органы.
Гликоген является основным источником энергии в организме. В норме гликоген хранится в печени до тех пор, пока организму не потребуется энергия. Затем ферменты превращают гликоген в глюкозу, чтобы она могла перемещаться по кровотоку к клеткам, которым требуется топливо.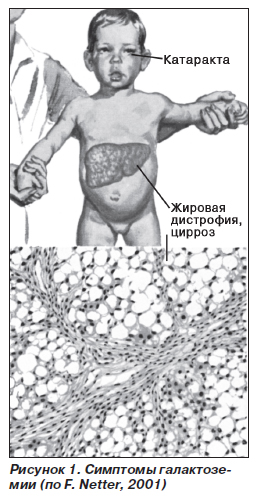 Каждая клетка тела содержит ферменты, но у детей с GSD отсутствует один из ферментов, отвечающих за выработку гликогена или превращение гликогена в глюкозу.
Каждая клетка тела содержит ферменты, но у детей с GSD отсутствует один из ферментов, отвечающих за выработку гликогена или превращение гликогена в глюкозу.
GSD — редкое заболевание. По данным Национальной организации редких заболеваний, GSD поражает менее 1 из 40 000 человек в Соединенных Штатах.
Какие существуют типы GSD?
Существует много различных типов GSD, в зависимости от того, какой фермент отсутствует. Некоторые типы поражают только печень, другие только мышцы, а некоторые поражают и печень, и мышцы. Каждый тип имеет немного разные симптомы. Лечение различается для различных типов GSD.
Наиболее распространенные типы GSD включают:
Болезнь накопления гликогена I типа (БГБ I) , также известная как болезнь фон Гирке, встречается примерно у 25 процентов всех детей с ГБ. Симптомы обычно появляются, когда ребенку от 3 до 4 месяцев, и могут включать гипогликемию (низкий уровень сахара в крови), которая может вызывать усталость, постоянный голод и раздражительность. Печень и иногда почки отекают из-за накопления гликогена.
Печень и иногда почки отекают из-за накопления гликогена.
Болезнь накопления гликогена III типа (GSD III) , также известная как болезнь Кори или болезнь Форбса, вызывает накопление гликогена в печени и мышцах. Симптомы обычно появляются в течение первого года жизни. У детей с этим типом GSD может быть вздутие живота, задержка роста и слабость мышц.
Болезнь накопления гликогена IV типа (GSD IV) , также известная как болезнь Андерсена, является одним из наиболее серьезных типов GSD. Симптомы обычно появляются в первый месяц жизни ребенка и включают неспособность набирать вес или расти с ожидаемой скоростью. Этот тип GSD часто приводит к циррозу печени, а также может поражать сердце и другие органы. Исходы ребенка зависят от формы GSD IV, которую они унаследовали.
Каковы риски GSD?
Каждый тип GSD несет определенные риски.
У младенцев с типом I (GSD I) может быть низкий уровень сахара в крови. Этот тип GSD также может привести к лактоацидозу, накоплению молочной кислоты, что может вызвать болезненные мышечные спазмы. По мере взросления у детей с GSD I может наблюдаться задержка полового созревания и слабость костей (остеопороз). Другие риски включают:
По мере взросления у детей с GSD I может наблюдаться задержка полового созревания и слабость костей (остеопороз). Другие риски включают:
- подагру, тип артрита
- аденомы, опухоли печени, обычно доброкачественные (нераковые)
- воспалительное заболевание кишечника (тип 1b)
- проблемы с зубами
- рецидивирующие инфекции (тип 1b)
- легочная гипертензия
Младенцы с типом III (GSD III) могут иметь низкий уровень сахара в крови и избыток жира в крови. По мере взросления их печень может увеличиваться. Печень некоторых детей возвращается к нормальному размеру, когда они достигают подросткового возраста, но у других развивается цирроз и печеночная недостаточность. Дети с этим типом GSD также подвержены риску:
- медленного роста и низкого роста
- мышечная слабость
У младенцев с типом IV (GSD IV) может не быть низкого уровня сахара в крови, но у них могут развиться ранние осложнения. Дети, выжившие с GSD IV, подвержены риску следующих осложнений:
- медленная прибавка в весе
- мышечная слабость, включая слабость сердечной мышцы
- цирроз
- портальная гипертензия
Болезнь накопления гликогена у детей
ИНФОРМАЦИЯ ПРИЧИНЫ ДИАГНОЗ УХОД СЛЕДУЮЩИЕ ШАГИ
Что такое болезнь накопления гликогена у детей?
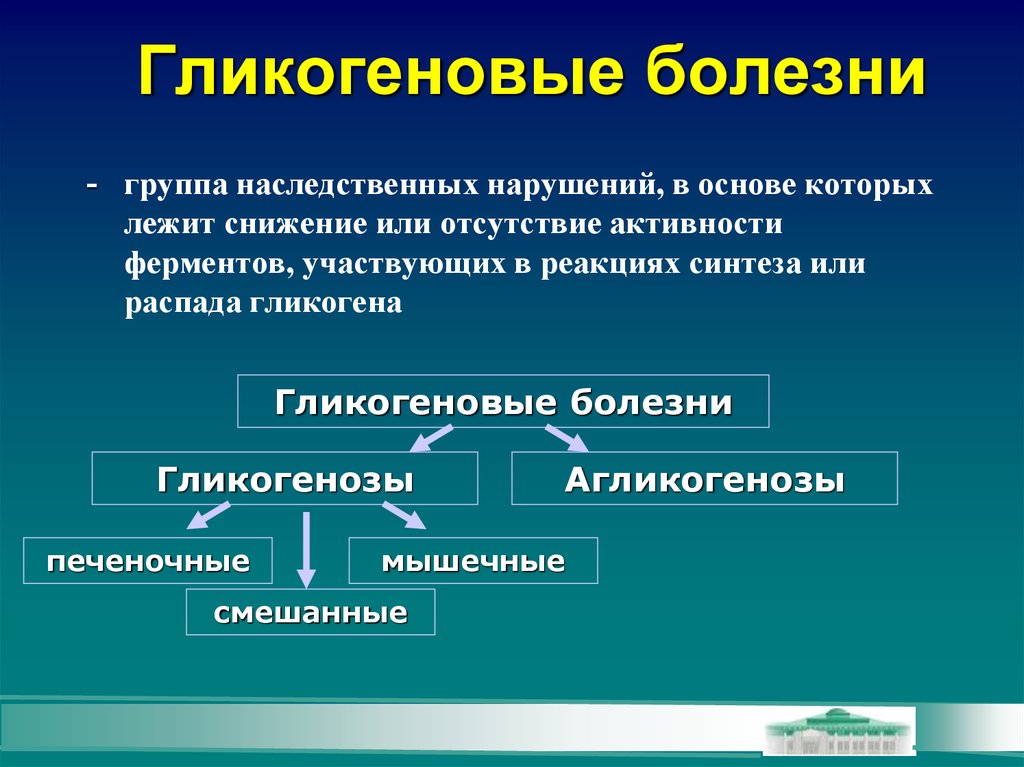
Болезнь накопления гликогена (GSD) — это редкое заболевание, которое изменяет способ использования и хранения гликогена, формы сахара или глюкозы.
Гликоген является основным источником энергии для организма. Гликоген хранится в печени. Когда организму требуется больше энергии, определенные белки, называемые ферментами, расщепляют гликоген на глюкозу. Они посылают глюкозу в организм.
Когда у кого-то есть GSD, у него отсутствует один из ферментов, расщепляющих гликоген. Когда фермент отсутствует, гликоген может накапливаться в печени. Или гликоген может не образовываться должным образом. Это может вызвать проблемы с печенью, мышцами или другими частями тела.
GSD передается от родителей к детям (наследственно). Чаще всего это наблюдается у младенцев или маленьких детей. Но некоторые формы GSD могут появиться и у взрослых.
Типы GSD
Специалистам известно не менее 9 типов GSD. Они сгруппированы по ферменту, который отсутствует в каждом из них. Каждый GSD имеет свои собственные симптомы и требует различного лечения.
Наиболее распространенными типами GSD являются типы I, III и IV:
- Тип I или Болезнь фон Гирке. Это наиболее распространенная форма GSD. У людей с типом I нет фермента, необходимого для превращения гликогена в глюкозу в печени. Гликоген накапливается в печени. Симптомы часто появляются у детей в возрасте от 3 до 4 месяцев. Они могут включать низкий уровень сахара в крови (гипогликемия) и вздутие живота из-за увеличенной печени.
- Тип III или Болезнь Кори. У людей с типом III недостаточно фермента (фермента разветвления), который помогает расщеплять гликоген. Гликоген не может полностью распасться. Он накапливается в печени и в мышечных тканях. Симптомы включают вздутие живота, задержку роста и слабость мышц.
- Тип IV или болезнь Андерсона. У людей с типом IV образуется аномальный гликоген. Эксперты считают, что аномальный гликоген запускает систему борьбы с инфекциями (иммунную систему). Это приводит к рубцеванию (циррозу) печени и других органов, таких как мышцы и сердце.
 Это наиболее распространенная форма GSD. У людей с типом I нет фермента, необходимого для превращения гликогена в глюкозу в печени. Гликоген накапливается в печени. Симптомы часто появляются у детей в возрасте от 3 до 4 месяцев. Они могут включать низкий уровень сахара в крови (гипогликемия) и вздутие живота из-за увеличенной печени.
Это наиболее распространенная форма GSD. У людей с типом I нет фермента, необходимого для превращения гликогена в глюкозу в печени. Гликоген накапливается в печени. Симптомы часто появляются у детей в возрасте от 3 до 4 месяцев. Они могут включать низкий уровень сахара в крови (гипогликемия) и вздутие живота из-за увеличенной печени. Это приводит к рубцеванию (циррозу) печени и других органов, таких как мышцы и сердце.
Это приводит к рубцеванию (циррозу) печени и других органов, таких как мышцы и сердце.Что вызывает болезнь накопления гликогена у ребенка?
Болезнь накопления гликогена передается от родителей к детям (наследственная).
Это происходит потому, что оба родителя имеют аномальный ген (генная мутация), который влияет на особый способ хранения или использования гликогена. Большинство GSD возникает из-за того, что оба родителя передают своим детям один и тот же аномальный ген.
В большинстве случаев родители не проявляют никаких симптомов заболевания.
Какие дети подвержены риску болезни накопления гликогена?
Болезнь накопления гликогена передается от родителей к детям (по наследству). Кто-то более подвержен риску GSD, если у него есть член семьи с этим заболеванием.
Каковы симптомы болезни накопления гликогена у ребенка?
При многих типах GSD симптомы сначала появляются у младенцев или у очень маленьких детей.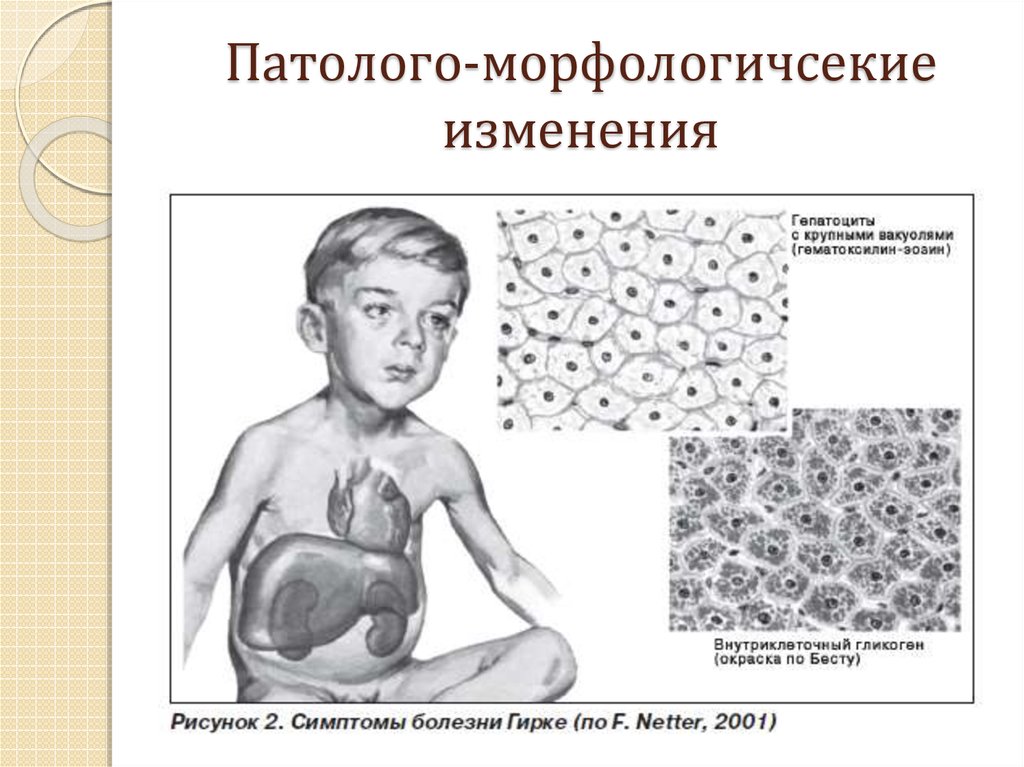 Симптомы будут варьироваться в зависимости от типа GSD у ребенка и от того, какой фермент отсутствует.
Симптомы будут варьироваться в зависимости от типа GSD у ребенка и от того, какой фермент отсутствует.
Поскольку GSD чаще всего поражает мышцы и печень, в этих областях проявляется больше всего симптомов.
Общие симптомы GSD могут включать:
- Недостаточно быстрый рост
- Некомфортное самочувствие в жаркую погоду (непереносимость жары)
- Слишком легко появляются синяки
- Низкий уровень сахара в крови (гипогликемия)
- Увеличение печени
- Вздутый живот
- Слабые мышцы (низкий мышечный тонус)
- Боль в мышцах и судороги во время упражнений
Симптомы у младенцев могут включать:
- Избыток кислоты в крови (ацидоз)
- Высокий уровень холестерина в крови (гиперлипидемия)
Симптомы GSD могут выглядеть как другие проблемы со здоровьем. Всегда посещайте лечащего врача вашего ребенка, чтобы быть уверенным.
Всегда посещайте лечащего врача вашего ребенка, чтобы быть уверенным.
Некоторые типы GSD могут проявляться у взрослых. Обратитесь к своему поставщику медицинских услуг, если вы думаете, что у вас может быть GSD.
Как диагностируется болезнь накопления гликогена у ребенка?
Лечащий врач вашего ребенка спросит о симптомах и состоянии здоровья вашего ребенка в прошлом. Медицинский работник проведет медицинский осмотр, чтобы проверить наличие таких симптомов, как увеличение печени или слабость мышц.
Врач вашего ребенка может сделать несколько анализов крови. Он или она может также взять небольшой образец ткани (биопсию) печени или мышц вашего ребенка. Образец будет доставлен в лабораторию. Будет проверено, чтобы увидеть, сколько определенного фермента находится в этой части тела.
Если вы беременны и беспокоитесь о GSD, ваш лечащий врач может провести некоторые тесты до рождения вашего ребенка (пренатальные тесты) для проверки на GSD.
Как лечить болезнь накопления гликогена у ребенка?
Лечение зависит от того, какой тип GSD у вашего ребенка.
При типах I, III и IV лечащий врач вашего ребенка может порекомендовать специальную диету, помогающую контролировать симптомы. Возможно, вашему ребенку также придется принимать определенные лекарства.
При других типах GSD вашему ребенку может потребоваться ограничить физические нагрузки, чтобы избежать мышечных спазмов. Ему или ей может потребоваться лечение для замены отсутствующего фермента (ферментозаместительная терапия).
Каковы осложнения болезни накопления гликогена у ребенка?
Накопление гликогена может повредить печень и мышцы. Это может создать другие проблемы, если у вашего ребенка есть определенные типы GSD, такие как:
- Тип III. Это может вызвать безвредные (доброкачественные) опухоли в печени.
- Тип IV. Со временем это может вызвать рубцевание (цирроз) печени. Это заболевание приводит к печеночной недостаточности.
Что я могу сделать, чтобы предотвратить болезнь накопления гликогена у моего ребенка?
Невозможно предотвратить болезнь накопления гликогена. Но раннее лечение может помочь контролировать симптомы, если у ребенка есть GSD.
Но раннее лечение может помочь контролировать симптомы, если у ребенка есть GSD.
Если у вас или вашего партнера есть GSD или семейный анамнез этого заболевания, обратитесь к консультанту-генетику, прежде чем забеременеть. Он или она может узнать ваши шансы на рождение ребенка с GSD.
Как я могу помочь моему ребенку жить с болезнью накопления гликогена?
У ребенка с GSD могут быть особые потребности. Убедитесь, что ваш ребенок получает регулярную медицинскую помощь. Важно, чтобы лечащий врач проверил состояние вашего ребенка. Регулярные визиты к врачу также помогут вам быть в курсе новых вариантов лечения.
Онлайн-группы или очные группы поддержки также могут быть полезны для вас и вашей семьи.
Когда мне следует звонить лечащему врачу моего ребенка?
Многие формы болезни накопления гликогена проявляются у младенцев и детей младшего возраста.
Позвоните своему лечащему врачу, если поведение вашего ребенка изменится после прекращения ночных кормлений.
Поговорите со своим лечащим врачом, если ваш ребенок:
- Растет недостаточно быстро
- Имеет постоянный (хронический) голод
- Вздулся живот
Подростки и взрослые должны обращать внимание на следующие симптомы во время физических упражнений:
- Мышечная слабость
- Боль
- Судороги
Основные сведения о болезни накопления гликогена у детей
- Болезнь накопления гликогена (БГН) — это редкое заболевание, которое изменяет способ использования и хранения организмом гликогена, формы сахара.
- Передается от родителей к детям (по наследству). Для большинства GSD каждый родитель должен передать одну аномальную копию одного и того же гена.
- У большинства родителей нет признаков GSD.
- Существует по крайней мере 9 известных типов GSD. Каждый GSD имеет свои собственные симптомы и требует различного лечения.
- Симптомы часто впервые появляются у младенцев или детей младшего возраста. В некоторых случаях GSD может появиться у взрослых.
 Каждый GSD имеет свои собственные симптомы и требует различного лечения.
Каждый GSD имеет свои собственные симптомы и требует различного лечения.Дальнейшие действия
Советы, которые помогут вам получить максимальную отдачу от посещения поставщика медицинских услуг вашего ребенка:
- Знайте причину визита и то, что вы хотите, чтобы произошло.
- Перед визитом запишите вопросы, на которые вы хотите получить ответы.
- При посещении запишите название нового диагноза и любые новые лекарства, методы лечения или тесты. Также запишите все новые инструкции, которые ваш поставщик дает вам для вашего ребенка.
- Знайте, почему прописывается новое лекарство или лечение и как оно поможет вашему ребенку. Также знайте, каковы побочные эффекты.
- Спросите, можно ли лечить состояние вашего ребенка другими способами.