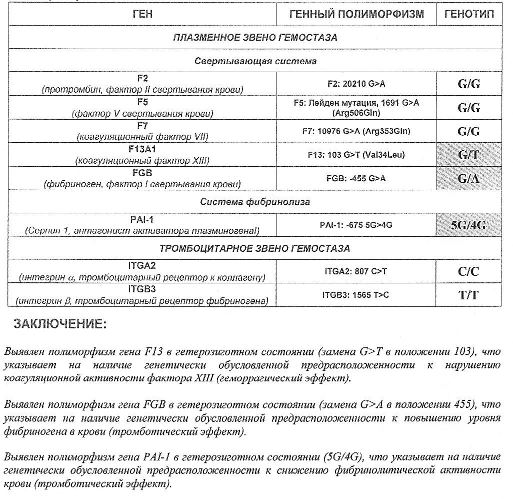
Мутации генов гемостаза цена: Расширенное исследование генов системы гемостаза (без описания результатов врачом-генетиком)
Расширенное исследование генов системы гемостаза (без описания результатов врачом-генетиком)
Метод определения Real-time-PCR.
Исследуемый материал Цельная кровь (с ЭДТА)
Доступен выезд на дом
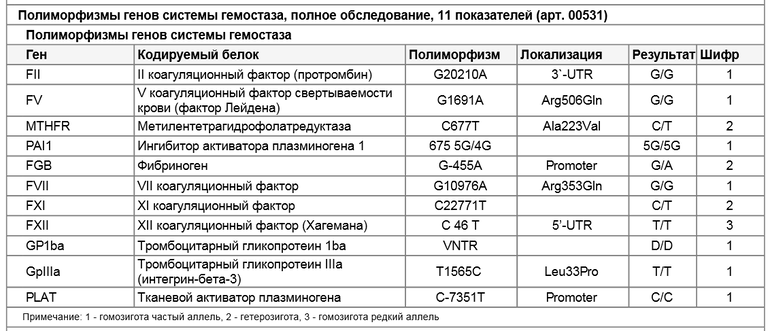
Расширенное исследование генов системы гемостаза: F2, F5, MTHFR, MTR, MTRR, F13, FGB, ITGA2, ITGВ3, F7, PAI-1
Комплексное исследование генетических факторов риска развития нарушений в системе свертывания крови и фолатном цикле (без заключения врача-генетика).
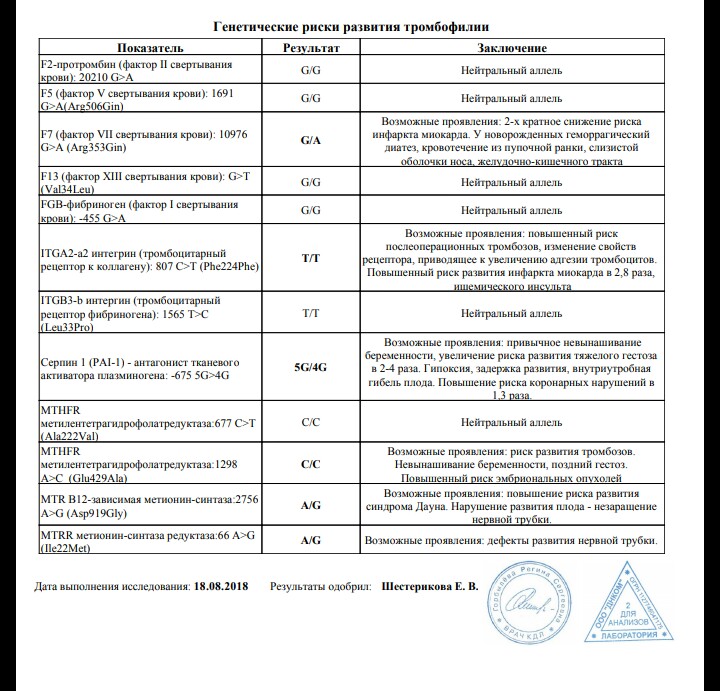
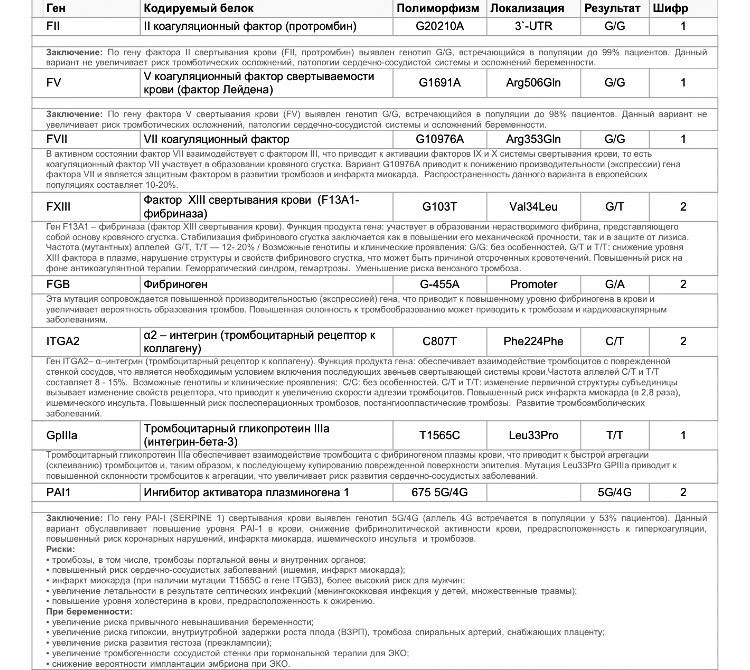
Различные изменения в генах системы гемостаза и цикла обмена фолатов предрасполагают к развитию большого числа патологических состояний: инфаркты, инсульты, тромбоэмболии, кровотечения, патология беременности и родов, осложнения послеоперационного периода и т.д. Профиль включает в себя исследование основных полиморфизмов в генах системы гемостаза и фолатного цикла:
- F2 c.
 *97G>A (20210 G>A; rs1799963),
*97G>A (20210 G>A; rs1799963), - F5 c.1601G>A (Arg534Gln; 1691 G>A; rs6025),
- MTHFR c.665C>T (Ala222Val; 677 C>T; rs1801133),
- MTHFR c.1286A>C (Glu429Ala; 1298 A>C; rs1801131),
- MTR c.2756A>G (Asp919Gly; rs1805087),
- MTRR c.66A>G (Ile22Met; rs1801394),
- F13 с.103G>T (I63Т; rs5985),
- FGB c.-467G>A (-455 G>А; rs1800790),
- ITGA2 c.759C>T (Phe253Phe, 807 C>T; rs1126643),
- ITGB3 c.176T>C (Leu59Pro; 1565 T>C; rs5918),
- F7 c.1238G>A (Arg353Gln; 10976 G>A; rs6046),
- PAI-1 (SERPINE1) –675 5G>4G (rs1799889).
 *97G>A (20210 G>A; rs1799963),
*97G>A (20210 G>A; rs1799963),
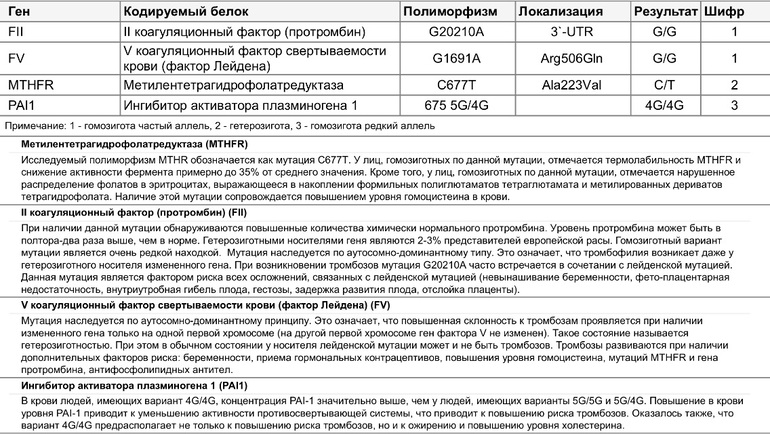
Ген F2 кодирует аминокислотную последовательность белка протромбина. Полиморфизм F2 c.*97G>A приводит к повышенной экспрессии гена. Клинически неблагоприятный вариант полиморфизма (c.*97A) наследуется по аутосомно-доминантному типу. Наличие полиморфизма F2 c.*97G>A в гомозиготной или гетерозиготной форме значительно (в 3 и более раз, а на фоне курения — в 40 и более раз) увеличивает риск возникновения венозных тромбозов, в том числе тромбозов сосудов мозга и сердца, особенно в молодом возрасте.
Ген F5 кодирует аминокислотную последовательность белка проакцелерина — коагуляционного фактора 5. Нуклеотидная замена c.1601G>A («мутация Лейден») приводит к аминокислотной замене аргинина на глутамин в позиции 534, что придает устойчивость активной форме проакцелерина. Клинически это проявляется рецидивирующими венозными тромбозами и тромбоэмболиями. Наличие полиморфизма в гомозиготной или гетерозиготной форме значительно (в 3 и более раз, а на фоне заместительной гормонотерапии или приема оральных контрацептивов — в 30 и более раз) увеличивает риск венозных тромбозов. Риск инфаркта миокарда увеличивается в 2 и более раз, риск развития патологии беременности (прерывание беременности, преэклампсия, хроническая плацентарная недостаточность и синдром задержки роста плода) увеличивается в 3 и более раз.
Также, пациенты, являющиеся одновременно носителями полиморфизма c.*97G>A гена протромбина и «мутации Лейден», еще в большей степени подвержены риску развития тромбозов и тромбоэмболий.
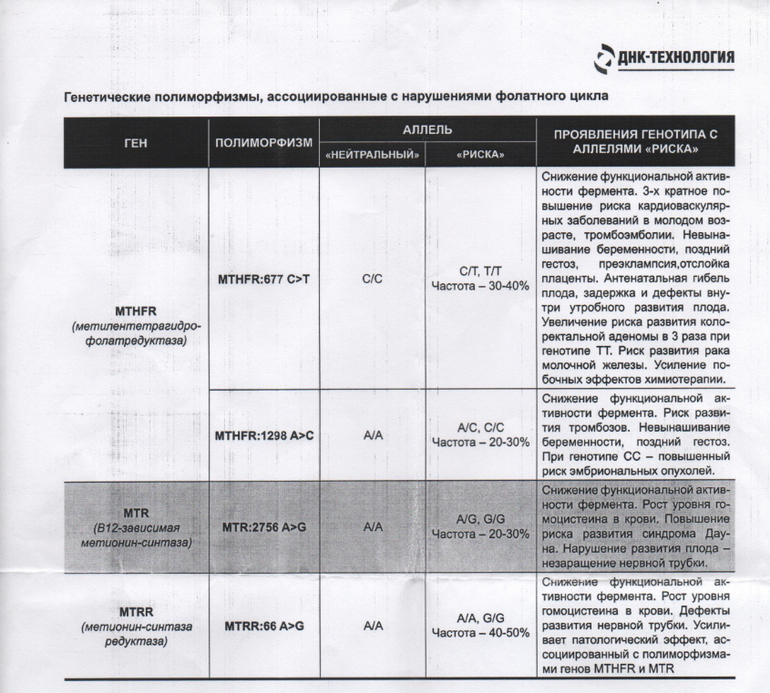
Ген MTHFR кодирует аминокислотную последовательность фермента метилентетрагидрофолатредуктазы, играющего ключевую роль в метаболизме фолиевой кислоты. Полиморфизм c.665C>T гена MTHFR связан с заменой нуклеотида цитозина (С) на тимин (Т), что приводит к аминокислотной замене аланина на валин в позиции 222. Вариант c.665Т связан с четырьмя группами мультифакториальных заболеваний: сердечно-сосудистыми, дефектами развития плода, колоректальной аденомой и раком молочной железы и яичников. У женщин с генотипом c.665Т/Т дефицит фолиевой кислоты во время беременности может приводить к порокам развития плода, в том числе незаращению нервной трубки. Неблагоприятное воздействие варианта c.665Т- зависит от внешних факторов: низкого содержания в пище фолатов, курения, приема алкоголя.
Полиморфизм MTHFR c.1286A>C связан с точечной заменой нуклеотида аденина (А) на цитозин (С), что приводит к замене аминокислотного остатка глутаминовой кислоты на аланин в позиции 429, относящейся к регулирующей области молекулы фермента. При наличии данного полиморфизма отмечается снижение активности фермента MTHFR. Это снижение обычно не сопровождается изменением уровня гомоцистеина в плазме крови у носителей дикого варианта полиморфизма c.665C>T, однако сочетание аллельного варианта* c.1286C с аллелем c.665T приводит к снижению уровня фолиевой кислоты и соответствует по своему эффекту гомозиготному состоянию MTHFR c.665Т/T. При этом риск развития дефектов нервной трубки повышается в 2 раза. Жизнеспособность плодов, имеющих одновременно оба полиморфных варианта, также снижена.
Ген MTRR кодирует аминокислотную последовательность фермента редуктазы метионинсинтазы. Полиморфизм c.66A>G связан с аминокислотной заменой в молекуле фермента. В результате этой замены функциональная активность фермента снижается, что приводит к повышению риска развития дефектов нервной трубки у плода. Влияние полиморфизма усугубляется дефицитом витамина В12. При сочетании полиморфизма c.66A>G гена MTRR с полиморфизмом c.665C>T в гене MTHFR риск spina bifida увеличивается. Полиморфизм c.
Ген фибриназы (F13) кодирует синтез трансглютаминазы, участвующей в стабилизации фибринового сгустка и в формировании соединительной ткани. Аллельные варианты с.103G/Т и с.103Т/Т приводят к снижению уровня трансглютаминазы с образованием сетчатой структуры фибрина с более тонкими волокнами, меньшими порами, и изменением характеристик проникновения, которое в сочетании с другими факторами риска ассоциируется с возможным риском внутричерепных кровоизлияний и кровотечений из внутренних органов, а также привычным невынашиванием беременности. При этом аллельный вариант с.103Т может выступать в роли протективного фактора в отношении инфаркта миокарда и венозных тромбозов.
Ген FGB кодирует β-цепь фибриногена, являющегося предшественником фибрина. Аллельный вариант c.-467А обусловливает усиленную транскрипцию гена и может приводить к увеличению уровня фибриногена в крови и повышению вероятности образования тромбов при наличии дополнительных факторов риска.
Ген гликопротеина Gp1a (ITGA2) кодирует синтез альфа-2-субъединицы интегринов – специализированных рецепторов тромбоцитов. Аллельный вариант c.759Т вызывает изменение первичной структуры субъединицы и свойств рецепторов. При гетерозиготном (c.759C/T) варианте отмечается увеличение скорости адгезии тромбоцитов к коллагену I типа, что может приводить к повышенному риску тромбофилии, инфаркта миокарда и других сердечно-сосудистых заболеваний. Аллельный вариант c.759Т связывают со случаями резистентности к аспирину. Помимо этого, при гомозиготном (c.759Т/T) варианте значительно увеличивается количество рецепторов на поверхности тромбоцитов. В совокупности, при гомозиготном варианте данного полиморфизма значительно повышен риск тромбофилии, инфаркта миокарда и развития других острых эпизодов тромбообразования в возрасте до 50 лет, даже по сравнению с гетерозиготным вариантом.
Ген гликопротеина Gp3a (ITGB3) кодирует синтез бета-3 цепи интегринового комплекса GP2b\3a, участвующего в разнообразных межклеточных взаимодействиях (адгезии и сигнализации). Аллельный вариант c.176С (гетерозигота c.176T/C) обусловливает повышенную адгезию тромбоцитов и может приводить к увеличению риска развития острого коронарного синдрома, а также связан с синдромом привычного невынашивания беременности. Гомозиготный вариант c.176С/C обусловливает повышенную адгезию тромбоцитов и может приводить к значительному увеличению риска развития острого коронарного синдрома в возрасте до 50 лет. У лиц с полиморфными аллельными вариантами часто отмечается пониженная эффективность аспирина.
Аллельный вариант c.1238A (гетерозигота c.1238G/A и гомозигота c.1238А/A) гена F7 приводит к понижению экспрессии гена и снижению уровня фактора 7 в крови, рассматривается как протективный маркёр в отношении развития тромбозов и инфаркта миокарда.
Ген ингибитора активатора плазминогена (PAI-1) кодирует белок-антагонист тканевого и урокиназного активатора плазминогена. Преобладающим в популяции вариантом исследуемого полиморфизма является гетерозиготный вариант -675 5G/4G. В связи с этим данный полиморфизм самостоятельного диагностического значения не имеет, эффект возможно оценить в сочетании с другими факторами предрасполагающими к развитию патологии (например в сочетании с FGB c.-467A). Аллельный вариант -675 4G сопровождается большей активностью гена, чем -675 5G, что обусловливает более высокую концентрацию PAI-1 и уменьшение активности противосвёртывающей системы. Гомозигота -675 4G/4G ассоциирована с повышением риска тромбообразования, преэклампсии, нарушением функции плаценты и самопроизвольного прерывания беременности.
*Примечание: иногда в научной литературе при описании однонуклеотидных замен, характерных для генных полиморфизмов, встречается термин «мутантный аллель». Это терминологическая неточность, так как в классической генетике термин «мутантный аллель» традиционно рассматривается как синоним термина «мутация». При мутациях, как известно, изменение структуры гена приводит к образованию (экспрессии) нефункциональных белков и к неизбежному развитию наследственного заболевания. При полиморфизмах изменение в структуре гена приводит лишь к появлению белков с немного изменёнными физико-химическими свойствами. Такие изменения, как известно, проявляют себя при воздействии на организм различных факторов внешней среды или при изменении функционального состояния организма человека. И только в таких ситуациях функционирование белков со структурными особенностями может, либо способствовать ускорению развития заболевания, либо, напротив, тормозить формирование патологических процессов. Поэтому, на наш взгляд, для разграничения изменений в генах столь очень похожих структурно, но приводящих к несоизмеримо разным последствиям для организма, корректнее в отношении генных полиморфизмов применять понятие «аллельный вариант гена», а не «мутантный аллель».
Это терминологическая неточность, так как в классической генетике термин «мутантный аллель» традиционно рассматривается как синоним термина «мутация». При мутациях, как известно, изменение структуры гена приводит к образованию (экспрессии) нефункциональных белков и к неизбежному развитию наследственного заболевания. При полиморфизмах изменение в структуре гена приводит лишь к появлению белков с немного изменёнными физико-химическими свойствами. Такие изменения, как известно, проявляют себя при воздействии на организм различных факторов внешней среды или при изменении функционального состояния организма человека. И только в таких ситуациях функционирование белков со структурными особенностями может, либо способствовать ускорению развития заболевания, либо, напротив, тормозить формирование патологических процессов. Поэтому, на наш взгляд, для разграничения изменений в генах столь очень похожих структурно, но приводящих к несоизмеримо разным последствиям для организма, корректнее в отношении генных полиморфизмов применять понятие «аллельный вариант гена», а не «мутантный аллель».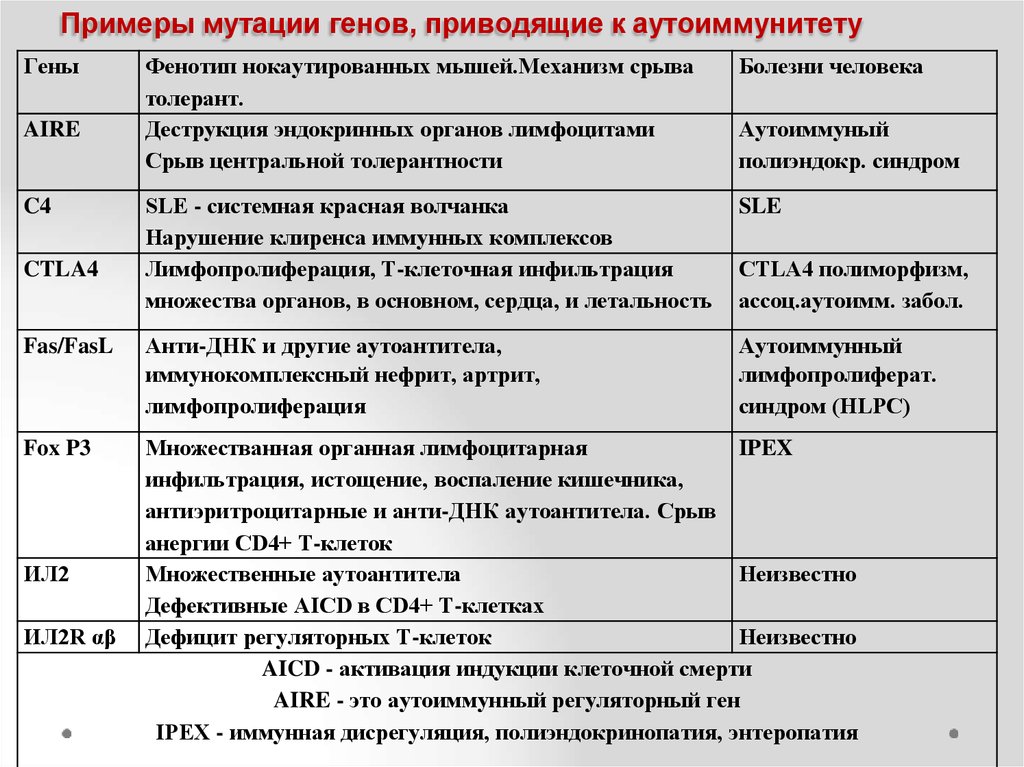
Мутации генов системы гемостаза. Анализ в Лаборатории CMD
Версия для печати
Биоматериал
Для данного исследования лаборатория принимает следующий биоматериал:
- Кровь с ЭДТА
Подготовка к исследованию
Не менее 3 часов после последнего приема пищи. Можно пить воду без газа.
Метод исследования
- ПЦР с детекцией в режиме «реального времени»
В случае же если для исследования полиморфизмов в генах F2 и F5 важен метод пиросеквенирование, то рекомендуется заказывать профиль 180014.
Склонность к повышенной коагуляции и формированию тромбов (тромбофилия) — глобальная медико-социальная проблема, основная причина смертности и инвалидизации во многих развитых странах мира. Частота венозных тромбозов в общей популяции, согласно мировым данным, составляет 1-2 случая на 1000 человек ежегодно.
В настоящее время хорошо изучены различные формы тромбофилии, выявлена наследственная составляющая заболевания и установлены причины заболевания на молекулярно-генетическом уровне.
Наиболее значимыми и часто встречающимися наследственными дефектами в системе гемостаза, приводящими к тромбофилии, являются полиморфизмы в генах, кодирующих коагуляционный фактор 5 (F5) и коагуляционный фактор 2 (F2, протромбин). Наличие одновременно двух полиморфизмов повышает риск тромбоза почти в 100 раз.
Также носительство данных полиморфизмов увеличивает вероятность развития гестоза, фетоплацентарной недостаточности, отставания развития плода, мертворождения. Существуют данные о повышении частоты встречаемости этих полиморфизмов у женщин с привычными выкидышами, особенно во II триместре беременности. Полиморфизмы достоверно ассоциированы с ранним и поздним привычным невынашиванием. Один из факторов риска развития тромбофилии у носителей полиморфизмов — прием комбинированных оральных контрацептивов.
Существуют данные о повышении частоты встречаемости этих полиморфизмов у женщин с привычными выкидышами, особенно во II триместре беременности. Полиморфизмы достоверно ассоциированы с ранним и поздним привычным невынашиванием. Один из факторов риска развития тромбофилии у носителей полиморфизмов — прием комбинированных оральных контрацептивов.
Показания к исследованию:
- Планирование любых оперативных вмешательств.
- Рецидивирующие венозные тромбозы.
- Тромбозы в молодом возрасте (до 40 лет).
- Варикозная болезнь вен нижних конечностей.
- Прегравидарная подготовка.
- Тромбоэмболические осложнения при беременности или на фоне приема комбинированных оральных контрацептивов.
- Женщины с отягощенным гинекологическим анамнезом (преждевременная отслойка нормально расположенной плаценты, плацентарная недостаточность, гестозы, невынашивание, мертворождение, неудачи ЭКО в анамнезе и т.п.).
- Беременные с сопуствующей экстрагенитальной патологией (ревматические пороки сердца, заболевания почек, артериальная гипертензия, метаболический синдром, воспалительные процессы различной локализации и т. п.).
- Беременные старшей возрастной группы (более 35 лет) с индуцированной беременностью, многоплодием.
 п.).
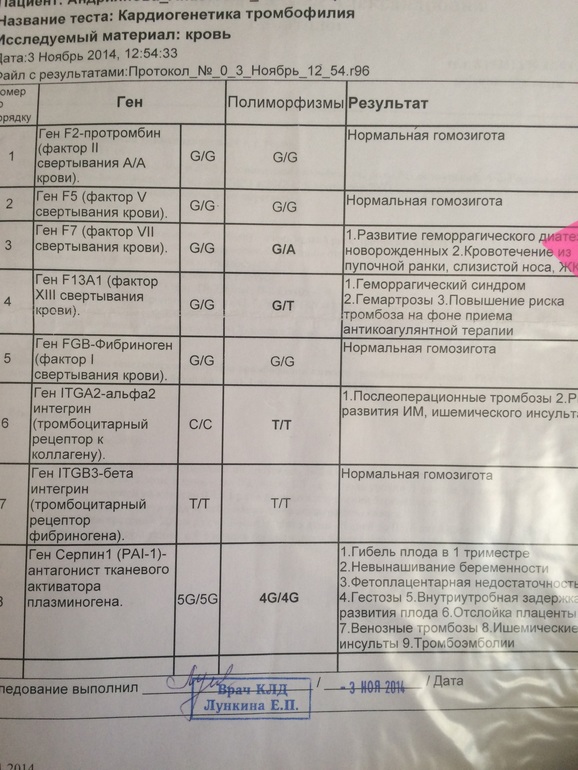
п.).Пример результата исследования.
| Параметр | Результат |
|---|---|
| Полиморфизм в гене F2 — rs1799963 (20210 G>A) | GG |
| Полиморфизм в гене F5 — мутация Лейден rs6025 (Arg506Gln, G>A) | GG |
|
Комментарий к генетическому исследованию: Изменения в последовательности ДНК могут приводить к нарушению закодированной в ней информации и проявлению соответствующих клинических признаков. Для интерпретации результатов необходима консультация врача-специалиста.
Результат лабораторного исследования не является диагнозом. Тактика обследования, лечения пациента, интерпретация результатов лабораторных исследований определяется лечащим врачом. |
|

Внимание!
- При необходимости по результатам исследований оформляется заключение врачом-генетиком (код услуги 181010).
- Заключение врача-генетика проводится только для услуг, выполняемых в лаборатории CMD.
- Врач-генетик описывает результат в течение 5 календарных дней после готовности генетического исследования
Обращаем Ваше внимание на то, что интерпретация результатов исследований, установление диагноза, а также назначение лечения, в соответствии с Федеральным законом № 323-ФЗ «Об основах охраны здоровья граждан в Российской Федерации» от 21 ноября 2011 года, должны производиться врачом соответствующей специализации.
- Код:
-
180010
можно сдать на дому
- Стоимость:
-
При единовременном заказе нескольких услуг, услуга по сбору биоматериала оплачивается только один раз.
2 563 р.
- + 190 р. Взятие крови
- Срок выполнения:
-
Указанный срок не включает день взятия биоматериала.
5-7 к.
д.
 д.
д.С этим анализом заказывают
Система гемостаза (скрининг)
- результаты за 3-6 часов (CITO)
- Код:
- 300006
- Срок:
- 1 к.д.
Цена: 1877 р.
Общий анализ крови + СОЭ с лейкоцитарной формулой (с микроскопией мазка крови при наличии патологических сдвигов), венозная кровь
- результаты за 3-6 часов (CITO)
- Код:
- 110006
- Срок:
- 1 к. д.
 д.
д.Цена: 743 р.
Антитела к кардиолипину (Anticardiolipin antibodies) класса IgG
- Код:
- 060734
- Срок:
- 1-2 к.д.
Цена: 1150 р.
Гомоцистеин (Homocysteine)
- результаты за 3-6 часов (CITO)
- Код:
- 060109
- Срок:
- 1 к.д.
Цена:
1821 р.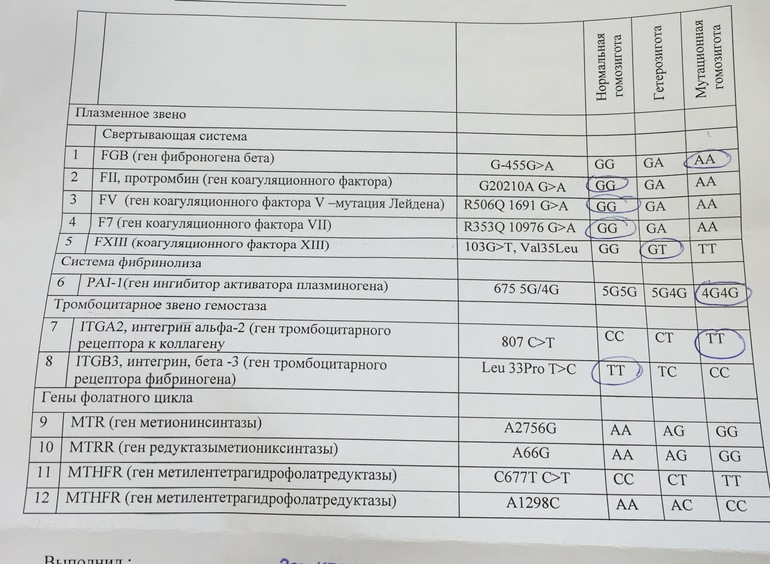
Ферритин (Ferritin)
- результаты за 3-6 часов (CITO)
- Код:
- 090031
- Срок:
- 1 к.д.
Цена: 693 р.
Показать еще
О возможных противопоказаниях необходимо проконсультироваться со специалистом
Высокая мутационная гетерогенность и новые мутации в гене V фактора свертывания крови человека. Будущие перспективы дефицита фактора V с использованием рекомбинантных и передовых методов лечения
1. Табибиан С. , Шираванд Ю., Шамс М., Сафа М., Голами М.С., Хейдари Ф., Ахмади А., Рашидпанах Дж., Доргалалех А.А. Всесторонний обзор фактора свертывания V и врожденного дефицита фактора V. Семин. тромб. Хемост. 2019;45:523–543. doi: 10.1055/s-0039-1687906. [PubMed] [CrossRef] [Академия Google]
, Шираванд Ю., Шамс М., Сафа М., Голами М.С., Хейдари Ф., Ахмади А., Рашидпанах Дж., Доргалалех А.А. Всесторонний обзор фактора свертывания V и врожденного дефицита фактора V. Семин. тромб. Хемост. 2019;45:523–543. doi: 10.1055/s-0039-1687906. [PubMed] [CrossRef] [Академия Google]
2. Сантамария С., Реглиньска-Матвеев Н., Гиерула М., Камире Р.М., Кроули Дж.Т.Б., Лейн Д.А., Анстрём Дж. Фактор V обладает кофакторной антикоагулянтной активностью, направленной на раннюю фазу свертывания крови. Дж. Биол. хим. 2017; 292:9335–9344. doi: 10.1074/jbc.M116.769570. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
3. Ван Дорн П., Розинг Дж., Дакерс С., Хакенг Т., Симиони П., Кастольди Э. Фактор V обладает антикоагулянтной активностью в плазме в присутствии TFPIα: разница между FV1 и FV2. тромб. Гемост. 2018;118:1194–1202. doi: 10.1055/s-0038-1656549. [PubMed] [CrossRef] [Google Scholar]
4. Segers K., Dahlbäck B., Nicolaes G. Коагуляционный фактор V и тромбофилия: предпосылки и механизмы.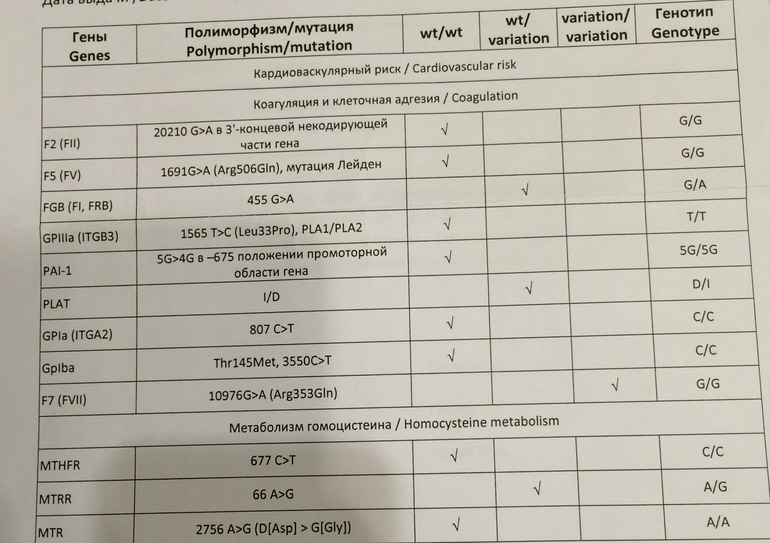 тромб. Гемост. 2007; 98: 530–542. doi: 10.1160/TH07-02-0150. [PubMed] [CrossRef] [Google Scholar]
тромб. Гемост. 2007; 98: 530–542. doi: 10.1160/TH07-02-0150. [PubMed] [CrossRef] [Google Scholar]
5. Hirbawi J., Vaughn J.L., Bukys M.A., Vos H.L., Kalafatis M. Вклад аминокислотного участка 659-663 тяжелой цепи фактора Va в активность фактора Xa внутри протромбиназы. Биохимия. 2010;49: 8520–8534. doi: 10.1021/bi101097t. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
6. Camire R.M. Новый взгляд на фактор свертывания крови V. Curr. мнение Гематол. 2011;18:338–342. doi: 10.1097/MOH.0b013e3283497ebc. [PubMed] [CrossRef] [Google Scholar]
7. Мазуркевич-Писарек А., Плуценничак Г., Циах Т., Плуценничак А. Белок фактора VIII и его функция. Акта Биохим. пол. 2016; 63:11–16. doi: 10.18388/abp.2015_1056. [PubMed] [CrossRef] [Академия Google]
8. Vadivel K., Kumar Y., Bunce M.W., Camire R.M., Bajaj M.S., Bajaj S.P. Взаимодействие кислой области B-домена фактора V с ее основной областью и с TFPI/TFPI2: структурные выводы из исследований молекулярного моделирования.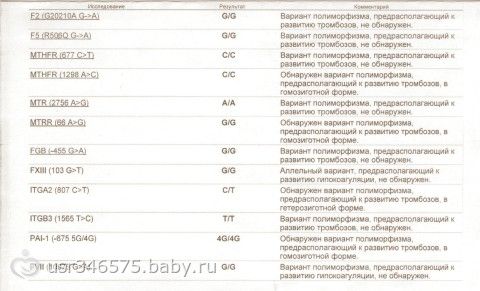 [(по состоянию на 2 сентября 2021 г.)]; Int. биол. Ред. 2017 г. 1 Доступно в Интернете: http://journals.ke-i.org/index.php/ibr/article/view/1334/975 [бесплатная статья PMC] [PubMed] [Google Scholar]
[(по состоянию на 2 сентября 2021 г.)]; Int. биол. Ред. 2017 г. 1 Доступно в Интернете: http://journals.ke-i.org/index.php/ibr/article/view/1334/975 [бесплатная статья PMC] [PubMed] [Google Scholar]
9. Bos M.H.A. , Камира Р.М. Факторы свертывания крови V и VIII: молекулярные механизмы активации прокофакторов. Дж. Коагул. Беспорядок. 2010;2:19–27. [Бесплатная статья PMC] [PubMed] [Google Scholar]
10. Ван Дорн П., Розинг Дж., Вилдерс С.Дж., Хакенг Т.М., Кастольди Э. С-конец ингибитора пути тканевого фактора-α ингибирует активацию фактора V с помощью Защита сайта расщепления Arg 1545 . Дж. Тромб. Гемост. 2017;15:140–149. doi: 10.1111/jth.13559. [PubMed] [CrossRef] [Google Scholar]
11. Фавалоро Э.Дж. Генетическое тестирование генов, связанных с тромбофилией: наблюдения за моделями тестирования фактора V Лейдена (G1691А) и «Мутация» гена протромбина (G20210A) Semin. тромб. Хемост. 2019;45:730–742. doi: 10.1055/s-0039-1694772. [PubMed] [CrossRef] [Google Scholar]
12. Рубен Э.А., Рау М.Дж., Фитцпатрик Дж., Ди Сера Э. Крио-ЭМ структуры факторов свертывания крови человека V и Va. Кровь. 2021;137:3137–3144. doi: 10.1182/blod.2021010684. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
Рубен Э.А., Рау М.Дж., Фитцпатрик Дж., Ди Сера Э. Крио-ЭМ структуры факторов свертывания крови человека V и Va. Кровь. 2021;137:3137–3144. doi: 10.1182/blod.2021010684. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
13. Хоффман М., Монро Д.А. Клеточная модель гемостаза. тромб. Гемост. 2001; 85: 958–965. doi: 10.1055/s-0037-1615947. [PubMed] [CrossRef] [Google Scholar]
14. Smith S.A. The Cell-Based Model of Coagulation. Дж. Вет. Эмердж. крит. Забота. 2009; 19:3–10. doi: 10.1111/j.1476-4431.2009.00389.x. [PubMed] [CrossRef] [Google Scholar]
15. Манн К.Г. протромбиназы: парадигма ферментных комплексов, связанных с мембраной; мемуары. Дж. Тромб. Тромболизис. 2021 г.: 10.1007/s11239-021-02402-w. [PubMed] [CrossRef] [Google Scholar]
16. Stoilova-McPhie S. Фактор VIII и фактор V Мембраносвязанные комплексы. В: Харрис Дж. Р., Марлес-Райт Дж., редакторы. Высокомолекулярные белковые комплексы III: структура и функции. Том 96. Международное издательство Спрингер; Чам, Швейцария: 2021. стр. 153–175. Субклеточная биохимия. [PubMed] [CrossRef] [Google Scholar]
стр. 153–175. Субклеточная биохимия. [PubMed] [CrossRef] [Google Scholar]
17. Kane W.H., Majerus P.W. Очистка и характеристика фактора свертывания крови человека V. J. Biol. хим. 1981; 256:1002–1007. doi: 10.1016/S0021-9258(19)70079-9. [PubMed] [CrossRef] [Google Scholar]
18. Gould W.R., Silveira J.R., Tracy P.B. Уникальные модификации фактора свертывания V in vivo производят физически и функционально отличный тромбоцитарный кофактор. Дж. Биол. хим. 2004;279: 2383–2393. doi: 10.1074/jbc.M308600200. [PubMed] [CrossRef] [Google Scholar]
19. Von Drygalski A., Bhat V., Gale A.J., Burnier L., Cramer T.J., Griffin J.H., Mosnier L.O. Созданный фактор Va предотвращает кровотечение, вызванное антикоагулянтом, активированным белком Wt C. PLoS ONE. 2014;9:e104304. doi: 10.1371/journal.pone.0104304. [Статья PMC бесплатно] [PubMed] [CrossRef] [Google Scholar]
20. Гейл А.Дж., Бхат В., Пеллекер Дж.Л., Гриффин Дж.Х., Мосниер Л.О., Фон Дрыгальский А. Безопасность, стабильность и фармакокинетические свойства SuperFactor Va, a Новый разработанный фактор свертывания крови V для лечения тяжелых кровотечений. фарм. Рез. 2016;33:1517–1526. дои: 10.1007/s11095-016-1895-3. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
фарм. Рез. 2016;33:1517–1526. дои: 10.1007/s11095-016-1895-3. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
21. Бхат В., фон Дрыгальский А., Гейл А.Дж., Гриффин Дж.Х., Монье Л.О. Улучшение коагуляции и гемостаза при гемофилии с помощью ингибиторов комбинаций суперфактора Va и фактора VIIa. тромб. Гемост. 2016; 115: 551–561. doi: 10.1160/th25-07-0525. [Статья бесплатно PMC] [PubMed] [CrossRef] [Google Scholar]
22. Cui J., O’Shea K.S., Purkayastha A., Saunders T.L., Ginsburg D. Фатальное кровотечение и неполный блок эмбриогенеза у мышей, лишенных фактора свертывания крови В. Природа. 1996;384:66–68. doi: 10.1038/384066a0. [PubMed] [CrossRef] [Google Scholar]
23. Yang T., Cui J., Taylor J., Yang A., Gruber S., Ginsburg D. Спасение фатального неонатального кровотечения у мышей с дефицитом фактора V с помощью низкого уровня Трансгенная экспрессия. тромб. Гемост. 2000;83:70–77. doi: 10.1055/s-0037-1613760. [PubMed] [CrossRef] [Google Scholar]
24. Weyand A.C., Grzegorski S.J., Rost M.S., Lavik K.I., Ferguson A.C., Menegatti M., Richter C.E., Asselta R., Duga S., Peyvandi F., et al. . Анализ фактора V у рыбок данио демонстрирует минимальные уровни, необходимые для раннего гемостаза. Кровь Adv. 2019;3:1670–1680. doi: 10.1182/bloodadvances.2018029066. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
Weyand A.C., Grzegorski S.J., Rost M.S., Lavik K.I., Ferguson A.C., Menegatti M., Richter C.E., Asselta R., Duga S., Peyvandi F., et al. . Анализ фактора V у рыбок данио демонстрирует минимальные уровни, необходимые для раннего гемостаза. Кровь Adv. 2019;3:1670–1680. doi: 10.1182/bloodadvances.2018029066. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
25. Cripe LD, Moore. К.Д., Кейн У.Х. Структура гена фактора свертывания крови человека V. Биохимия. 1992; 31: 3777–3785. doi: 10.1021/bi00130a007. [PubMed] [CrossRef] [Google Scholar]
26. Dall’Osso C., Guella I., Duga S., Locatelli N., Paraboschi E.M., Spreafico M., Afrasiabi A., Pechlaner C., Peyvandi F. , Tenchini M.L., et al. Молекулярная характеристика трех новых мутаций сплайсинга, вызывающих дефицит фактора V, и анализ паттерна сплайсинга гена F5. Гематология. 2008;93: 1505–1513. doi: 10.3324/гематол.12934. [PubMed] [CrossRef] [Google Scholar]
27. Guasch J.F., Cannegieter S., Reitsma P.H., Van ‘t Veer-Korthof E. T., Bertina R.M. Тяжелый дефицит фактора свертывания V, вызванный делецией 4 п.н. в гене фактора V: генетический дефект при тяжелом дефиците фактора V. бр. Дж. Гематол. 1998; 101:32–39. doi: 10.1046/j.1365-2141.1998.00664.x. [PubMed] [CrossRef] [Google Scholar]
T., Bertina R.M. Тяжелый дефицит фактора свертывания V, вызванный делецией 4 п.н. в гене фактора V: генетический дефект при тяжелом дефиците фактора V. бр. Дж. Гематол. 1998; 101:32–39. doi: 10.1046/j.1365-2141.1998.00664.x. [PubMed] [CrossRef] [Google Scholar]
28. База данных мутаций генов человека в Институте медицинской генетики в Кардиффе (HGMD) [(по состоянию на 2 сентября 2021 г.)]. Доступно в Интернете: http://www.hgmd.cf.ac.uk/ac/index.php
29. Caudill J.S., Sood R., Zehnder J.L., Pruthi R.K., Steensma D.P. Тяжелый дефицит фактора свертывания крови V, связанный с интерстициальной делецией хромосомы 1q. Дж. Тромб. Гемост. 2007; 5: 626–628. doi: 10.1111/j.1538-7836.2007.02363.x. [PubMed] [CrossRef] [Google Scholar]
30. Guella I., Paraboschi E.M., van Schalkwyk W.A., Asselta R., Duga S. Идентификация первой Alu-опосредованной большой делеции, затрагивающей ген F5, у сложного гетерозиготного пациента с тяжелым дефицитом фактора V. тромб. Гемост. 2011;106:296–303. doi: 10.1160/Th21-03-0149. [PubMed] [CrossRef] [Google Scholar]
doi: 10.1160/Th21-03-0149. [PubMed] [CrossRef] [Google Scholar]
31. Липпи Г., Фавалоро Э.Дж., Монтаньяна М., Манзато Ф., Гуиди Г.К., Франчини М. Унаследованный и приобретенный дефицит фактора V. Кровавый коагул. Фибринолиз. 2011;22:160–166. doi: 10.1097/MBC.0b013e3283424883. [PubMed] [CrossRef] [Google Scholar]
32. Талджи Н., Камира Р. Парагемофилия: новый взгляд на дефицит фактора V. Семин. тромб. Хемост. 2013; 39: 607–612. doi: 10.1055/s-0033-1349224. [PubMed] [CrossRef] [Академия Google]
33. Врожденный дефицит фактора V Портал редких заболеваний и орфанных препаратов Orphanet. [(по состоянию на 2 сентября 2021 г.)]. Доступно в Интернете: https://www.orpha.net/consor/cgi-bin/index.php?lng=EN
34. Mannucci P.M., Duga S., Peyvandi F. Рецессивно наследуемые нарушения коагуляции. Кровь. 2004; 104:1243–1252. doi: 10.1182/blood-2004-02-0595. [PubMed] [CrossRef] [Google Scholar]
35. Дженни Р.Дж., Питтман Д.Д., Тул Дж.Дж., Криз Р.В., Алдапе Р.А., Хьюик Р. М., Кауфман Р.Дж., Манн К.Г. Полная кДНК и производная аминокислотная последовательность человеческого фактора V. Proc. Натл. акад. науч. США. 1987;84:4846–4850. doi: 10.1073/pnas.84.14.4846. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
М., Кауфман Р.Дж., Манн К.Г. Полная кДНК и производная аминокислотная последовательность человеческого фактора V. Proc. Натл. акад. науч. США. 1987;84:4846–4850. doi: 10.1073/pnas.84.14.4846. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
36. Оурен П. Парагемофилия. Ланцет. 1947; 249: 446–448. doi: 10.1016/S0140-6736(47)91941-7. [PubMed] [CrossRef] [Google Scholar]
37. Милетич Дж.П., Майерус Д.В., Майерус П.В. У пациентов с врожденным дефицитом фактора V снижено количество сайтов связывания фактора Ха на тромбоцитах. Дж. Клин. расследование 1978; 62: 824–831. doi: 10.1172/JCI109194. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
38. Duckers C., Simioni P., Spiezia L., Radu C., Dabrilli P., Gavasso S., Rosing J., Castoldi E. Остаточный тромбоцитарный фактор V обеспечивает образование тромбина у пациентов с тяжелым врожденным дефицитом фактора V и легкие симптомы кровотечения. Кровь. 2010; 115: 879–886. doi: 10.1182/blood-2009-08-237719. [PubMed] [CrossRef] [Google Scholar]
[PubMed] [CrossRef] [Google Scholar]
39. Castoldi E., Duckers C., Radu C., Spiezia L., Rossetto V., Tagariello G., Rosing J., Simioni P. Homozygous F5 Deep-Intronic Мутация сплайсинга, приводящая к серьезному дефициту фактора V и неопределяемому образованию тромбина в богатой тромбоцитами плазме: глубокая интронная мутация, вызывающая тяжелый дефицит фактора V. Дж. Тромб. Гемост. 2011;9: 959–968. doi: 10.1111/j.1538-7836.2011.04237.x. [PubMed] [CrossRef] [Google Scholar]
40. Duckers C., Simioni P., Spiezia L., Radu C., Gavasso S., Rosing J., Castoldi E. Низкие плазменные уровни ингибитора пути тканевого фактора в Пациенты с врожденным дефицитом фактора V. Кровь. 2008; 112:3615–3623. doi: 10.1182/blod-2008-06-162453. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
41. Jain S., Acharya S.S. Лечение редких нарушений свертывания крови в 2018 г. Transfus. Афер. науч. 2018;57:705–712. doi: 10.1016/j.transci.2018.10.009. [PubMed] [CrossRef] [Google Scholar]
42. Peyvandi F., Di Michele D., Bolton-Maggs P.H.B., Lee C.A., Tripodi A., Srivastava A. Классификация редких нарушений свертывания крови (RBDs) на основе ассоциации между активностью коагулянтного фактора и клинической тяжестью кровотечения: классификация редких нарушений кровотечения. Дж. Тромб. Гемост. 2012; 10:1938–1943. doi: 10.1111/j.1538-7836.2012.04844.x. [PubMed] [CrossRef] [Google Scholar]
Peyvandi F., Di Michele D., Bolton-Maggs P.H.B., Lee C.A., Tripodi A., Srivastava A. Классификация редких нарушений свертывания крови (RBDs) на основе ассоциации между активностью коагулянтного фактора и клинической тяжестью кровотечения: классификация редких нарушений кровотечения. Дж. Тромб. Гемост. 2012; 10:1938–1943. doi: 10.1111/j.1538-7836.2012.04844.x. [PubMed] [CrossRef] [Google Scholar]
43. Gupta G.K., Hendrickson J.E., Bahel P., Siddon A.J., Rinder H.M., Tormey C.A. Активность фактора V в тромбоцитах после афереза: последствия для лечения дефицита FV. Переливание. 2021; 61: 405–409. doi: 10.1111/trf.16179. [PubMed] [CrossRef] [Google Scholar]
44. Джимальски Д.М., Эльсайес А.Х., Уорд К.Р., Хаус М., Маница В.С. Переливание тромбоцитов как лечение дефицита фактора V у роженицы: клинический случай. Переливание. 2019;59:2234–2237. doi: 10.1111/trf.15322. [PubMed] [CrossRef] [Google Scholar]
45. Heger A., Svae T.E., Neisser-Svae A., Jordan S., Behizad M.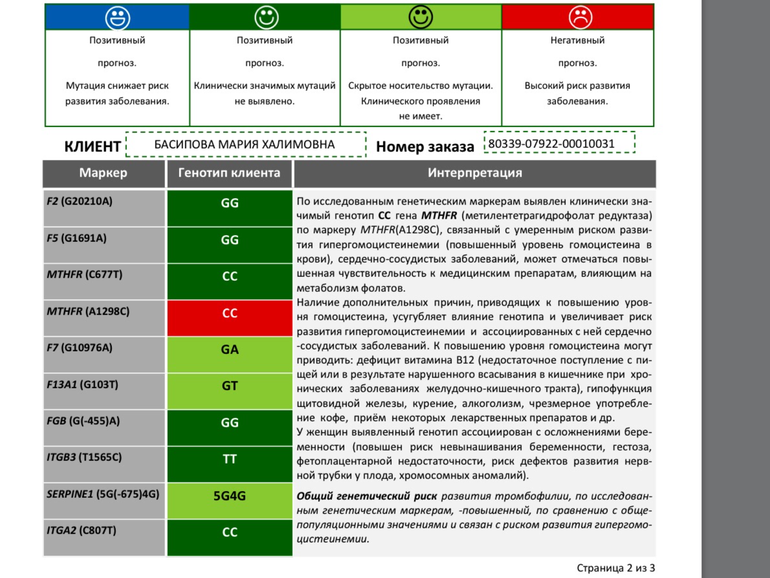 , Römisch J. Биохимическое качество фармацевтически лицензированной плазмы OctaplasLG ® после Внедрение нового прионного белка (PrP Sc ) Технология удаления и сокращение времени обработки растворителем/детергентом (S/D). Вокс Санг. 2009; 97: 219–225. doi: 10.1111/j.1423-0410.2009.01190.x. [PubMed] [CrossRef] [Google Scholar]
, Römisch J. Биохимическое качество фармацевтически лицензированной плазмы OctaplasLG ® после Внедрение нового прионного белка (PrP Sc ) Технология удаления и сокращение времени обработки растворителем/детергентом (S/D). Вокс Санг. 2009; 97: 219–225. doi: 10.1111/j.1423-0410.2009.01190.x. [PubMed] [CrossRef] [Google Scholar]
46. Cushing M.M., Asmis L., Calabia C., Rand J.H., Haas T. Эффективность плазмы с растворителем/детергентом после хранения при 2–8 °C в течение 5 дней в сравнении к другим продуктам плазмы для улучшения уровня фактора V в плазме с дефицитом фактора V. Трансфус. Афер. науч. 2016;55:114–119. doi: 10.1016/j.transci.2016.04.015. [PubMed] [CrossRef] [Академия Google]
47. Spinella P.C., Borasino S., Alten J. Плазма, обработанная растворителем/детергентом, в лечении педиатрических пациентов, которым требуется замена нескольких факторов свертывания крови: открытое, многоцентровое, постмаркетинговое исследование. Передний. Педиатр.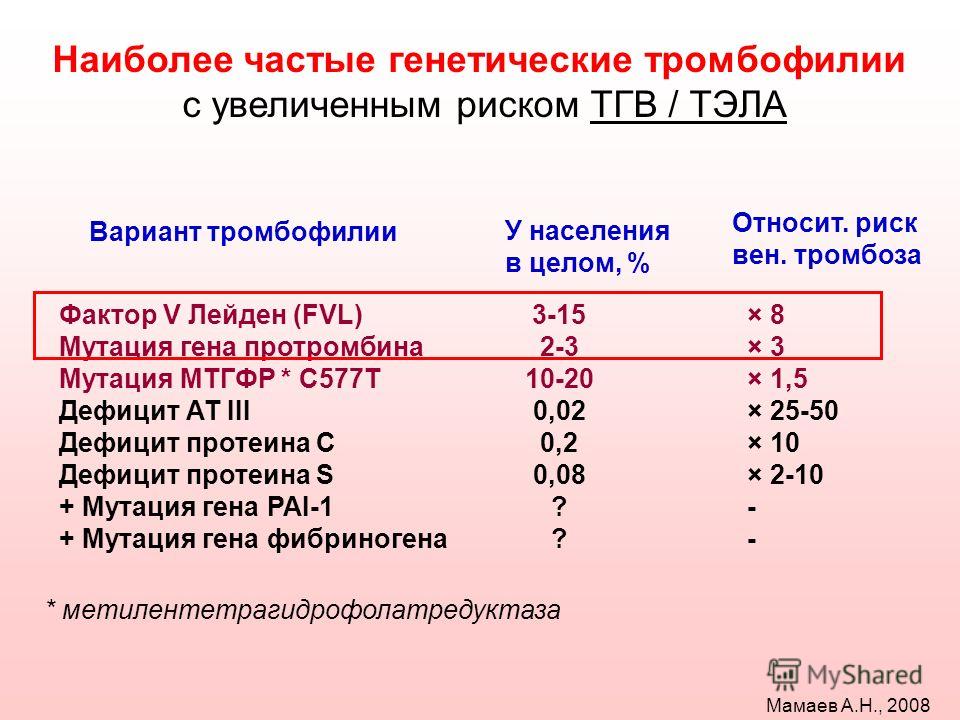 2020;8:572. doi: 10.3389/fped.2020.00572. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
2020;8:572. doi: 10.3389/fped.2020.00572. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
48. Гуленок Т., Васко С., Файл Д., Айзенберг Н., Де Рокур Э., Дюпон А., Фрер С., Джеймс С. ., Рабут Э., Ругери Л. и др. Учебная группа РАВИ. Приобретенный ингибитор фактора V: общенациональное исследование 38 пациентов. бр. Дж. Гематол. 2021;192: 892–899. doi: 10.1111/bjh.17308. [PubMed] [CrossRef] [Google Scholar]
49. Лунги Б., Кастольди Э., Мингоцци Ф., Бернарди Ф., Кастаман Г. Новая нулевая мутация фактора V, обнаруженная у пациента с тромбофилией и псевдогомозиготной резистентностью к APC и у бессимптомного неродственного субъекта. Кровь. 1998; 92: 1463–1464. doi: 10.1182/кровь.V92.4.1463. [PubMed] [CrossRef] [Google Scholar]
50. Патент «Метод in vitro для восстановления экспрессии гена F5, кодирующего фактор свертывания крови V». Патент на изобретение с экспертизой. Признание патента. Управление передачи результатов исследований. 2021. [(по состоянию на 2 сентября 2021 г. )]. Доступно в Интернете: https://consultas2.oepm.es/pdf/ES/0000/000/02/78/53/ES-2785323_B2.pdf
)]. Доступно в Интернете: https://consultas2.oepm.es/pdf/ES/0000/000/02/78/53/ES-2785323_B2.pdf
51. Метод in vitro для восстановления экспрессии гена F5, кодирующего фактор свертывания крови V. Каталог переноса. Управление передачи результатов исследований. 2021. [(по состоянию на 2 сентября 2021 г.)]. Доступно онлайн: https://www.ucm.es/otri/complutransfer-metodo-in-vitro-para-recuperar-la-expresion-del-gen-f5-que-codifica-el-factor-v-de-la -coagulacion-1
52. Ajzner E., Balogh I., Szabó T., Marosi A., Haramura G., Muszbek L. Серьезный дефицит фактора свертывания крови V, вызванный двумя новыми мутациями сдвига рамки считывания: 2952delT в экзоне 13 и 5493insG в экзоне 16 гена фактора 5. Кровь. 2002; 99: 702–705. doi: 10.1182/blood.V99.2.702. [PubMed] [CrossRef] [Google Scholar]
53. Махесвари К., Вадхва Л. Роль кровного родства в педиатрических неврологических расстройствах. Междунар. Дж. Контемп. Педиатрия. 2016;3:939–942. doi: 10.18203/2349-3291.ijcp20162369./9/9.jpg) [CrossRef] [Google Scholar]
[CrossRef] [Google Scholar]
54. Бхиндер М.А., Садия Х., Махмуд Н., Касим М., Хуссейн З., Рашид М.М., Захур М.Ю., Бхатти Р., Шехзад В., Варья А.М. и др. Родственное родство: благо или угроза на популяционном уровне? Анна. Гум. Жене. 2019;83:214–219. doi: 10.1111/ahg.12308. [PubMed] [CrossRef] [Google Scholar]
55. Ония О., Невес К., Ахмед Б., Конье Дж. К. Обзор репродуктивных последствий кровного родства. Евро. Дж. Обст. Гинекол. Воспр. биол. 2019; 232:87–96. doi: 10.1016/j.ejogrb.2018.10.042. [PubMed] [CrossRef] [Google Scholar]
56. Надери М., Табибиан С., Ализаде С., Хоссейни С., Закер Ф., Бамеди Т., Шамзизаде М., Доргалале А. Врожденный дефицит фактора V: Сравнение тяжести клинических проявлений у пациентов с редкими нарушениями свертываемости крови. Акта Гематол. 2015; 133:148–154. дои: 10.1159/000363598. [PubMed] [CrossRef] [Google Scholar]
57. Бхопал Р.С., Петерик Э.С., Райт Дж., Смолл Н. Потенциальные социальные, экономические и общие преимущества для здоровья кровнородственных браков: результаты когортного исследования «Рожденные в Брэдфорде». Евро. Дж. Общественное здравоохранение. 2014; 24:862–869. doi: 10.1093/eurpub/ckt166. [PubMed] [CrossRef] [Google Scholar]
Евро. Дж. Общественное здравоохранение. 2014; 24:862–869. doi: 10.1093/eurpub/ckt166. [PubMed] [CrossRef] [Google Scholar]
58. Castoldi E., Lunghi B., Mingozzi F., Muleo G., Redaelli R., Mariani G., Bernardi F. Мутация Missense (Y1702C) в коагуляции Ген фактора V является частой причиной дефицита фактора V у итальянского населения. Гематология. 2001;86:629–633. [PubMed] [Google Scholar]
59. Kong R.X., Xie Y.S., Xie H.X., Luo S.S., Wang M.S. Анализ фенотипа и генотипа семьи с наследственным дефицитом фактора свертывания крови V, вызванным сложной гетерозиготной мутацией. Чжунго Ши Янь Сюэ Е Сюэ За Чжи. 2020;28:2033–2038. doi: 10.19746/j.cnki.issn.1009-2137.2020.06.039. [PubMed] [CrossRef] [Google Scholar]
60. Асселта Р., Тенчини М.Л., Дуга С. Наследственные дефекты фактора свертывания крови V: геморрагическая сторона: фактор V и нарушения свертываемости крови. Дж. Тромб. Гемост. 2006; 4: 26–34. doi: 10.1111/j.1538-7836.2005.01590.х. [PubMed] [CrossRef] [Google Scholar]
61. Дуга С., Монтефуско М.С., Асселта Р., Мальковати М., Пейванди Ф., Сантагостино Э., Маннуччи П.М., Тенчини М.Л. Миссенс-мутация Arg2074Cys в домене C2 фактора V, вызывающая умеренно тяжелый дефицит фактора V: молекулярная характеристика путем экспрессии рекомбинантного белка. Кровь. 2003; 101: 173–177. doi: 10.1182/blod-2002-06-1928. [PubMed] [CrossRef] [Google Scholar]
Дуга С., Монтефуско М.С., Асселта Р., Мальковати М., Пейванди Ф., Сантагостино Э., Маннуччи П.М., Тенчини М.Л. Миссенс-мутация Arg2074Cys в домене C2 фактора V, вызывающая умеренно тяжелый дефицит фактора V: молекулярная характеристика путем экспрессии рекомбинантного белка. Кровь. 2003; 101: 173–177. doi: 10.1182/blod-2002-06-1928. [PubMed] [CrossRef] [Google Scholar]
62. Nuzzo F., Bulato C., Nielsen B.I., Lee K., Wielders S.J., Simioni P., Key N.S., Castoldi E. Характеристика явно синонимичных F5 Мутация, вызывающая аберрантный сплайсинг и дефицит фактора V. гемофилия. 2015;21:241–248. doi: 10.1111/hae.12554. [PubMed] [CrossRef] [Google Scholar]
63. Кунья М.Л.Р., Бахтиари К., Питер Дж., Маркварт Дж.А., Мейерс Дж.К.М., Мидделдорп С. Новая мутация в гене F5 (фактор V Амстердам), связанная с независимым кровотечением прокоагулянтной функции фактора V. Кровь. 2015; 125:1822–1825. doi: 10.1182/blood-2014-08-592733. [PubMed] [CrossRef] [Google Scholar]
64. Yamazaki T., Nicolaes GA, Sørensen K.W., Dahlbäck B. Молекулярная основа количественного дефицита фактора V, связанного с гаплотипом фактора V R2. Кровь. 2002; 100: 2515–2521. doi: 10.1182/кровь.V100.7.2515. [PubMed] [CrossRef] [Академия Google]
Yamazaki T., Nicolaes GA, Sørensen K.W., Dahlbäck B. Молекулярная основа количественного дефицита фактора V, связанного с гаплотипом фактора V R2. Кровь. 2002; 100: 2515–2521. doi: 10.1182/кровь.V100.7.2515. [PubMed] [CrossRef] [Академия Google]
65. Монтефуско М.К., Дуга С., Асселта Р., Мальковати М., Пейванди Ф., Сантагостино Э., Маннуччи П.М., Тенчини М.Л. Клиническая и молекулярная характеристика 6 пациентов с тяжелым дефицитом фактора свертывания крови V: расширение мутационного спектра гена фактора V и анализ недавно выявленных миссенс-мутаций in vitro. Кровь. 2003; 102:3210–3216. doi: 10.1182/blood-2003-03-0922. [PubMed] [CrossRef] [Google Scholar]
66. Chen T.Y., Lin T.M., Chen H.Y., Wu C.L., Tsao C.J. Gly39Мутация 2Cys Missense в домене A2 фактора V, вызывающая тяжелую недостаточность фактора V: молекулярная характеристика экспрессией рекомбинантного белка. тромб. Гемост. 2005; 93: 614–615. doi: 10.1055/s-0037-1616564. [PubMed] [CrossRef] [Google Scholar]
67. Liu HC, Shen M.C., Eng HL, Wang C.H., Lin T.M. Мутация Asp68His в домене A1 человеческого фактора V вызывает нарушение секреции и неэффективную транслокацию. гемофилия. 2014; 20:e318–e326. doi: 10.1111/hae.12476. [PubMed] [CrossRef] [Академия Google]
Liu HC, Shen M.C., Eng HL, Wang C.H., Lin T.M. Мутация Asp68His в домене A1 человеческого фактора V вызывает нарушение секреции и неэффективную транслокацию. гемофилия. 2014; 20:e318–e326. doi: 10.1111/hae.12476. [PubMed] [CrossRef] [Академия Google]
68. Фридманн А.П., Кутыченко А., Ву С., Фреденбург Дж.К., Вейц Дж.И., Гросс П.Л., Сюй П., Ни Ф., Ким П.Ю. Идентификация и характеристика сайта связывания фактора Va на фрагменте протромбина человека 2. Sci. Отчет 2019; 9: 2436. doi: 10.1038/s41598-019-38857-4. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
69. Пейванди Ф., Гараджола И., Янг Г. Прошлое и будущее гемофилии: диагностика, лечение и ее осложнения. Ланцет. 2016; 388:187–197. doi: 10.1016/S0140-6736(15)01123-X. [PubMed] [CrossRef] [Академия Google]
70. Zhang B., Zheng C., Zhu M., Tao J., Vasievich M.P., Baines A., Kim J., Schekman R., Kaufman R.J., Ginsburg D. Мыши с дефицитом LMAN1 Exhibit FV и дефицитом FVIII и накопление A1-антитрипсина в печени.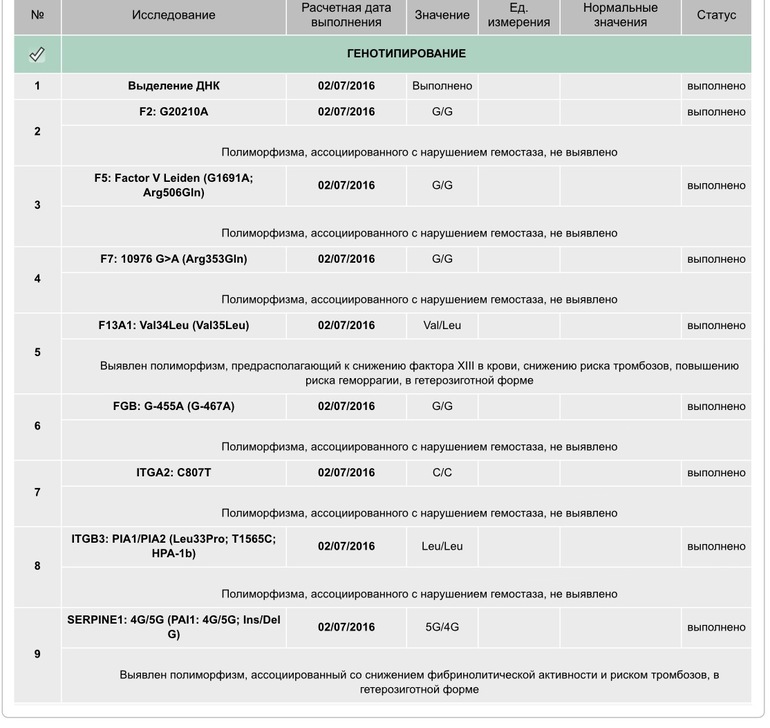 Кровь. 2011; 118:3384–3391. doi: 10.1182/blood-2011-05-352815. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
Кровь. 2011; 118:3384–3391. doi: 10.1182/blood-2011-05-352815. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
71. Дженкинс П. В., Дилл Дж. Л., Чжоу К., Фэй П. Дж. Вклад субъединиц фактора VIIIa A2 и A3-C1-C2 в сродство к фактору IXa у Фактор Хасэ. Биохимия. 2004;43:5094–5101. doi: 10.1021/bi036289p. [PubMed] [CrossRef] [Google Scholar]
72. ДеАнджелис Дж. П., Вакабаяши Х., Фэй П. Дж. Последовательности, фланкирующие Arg336 в факторе VIIIa, модулируют скорость катализируемого фактором Xa расщепления в этом сайте и функцию кофактора. Дж. Биол. хим. 2012; 287:15409–15417. doi: 10.1074/jbc.M111.333948. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
73. Williamson D., Brown K., Luddington R., Baglin C., Baglin T. Factor V Cambridge: A New Mutation (Arg306→Thr) Ассоциируется с устойчивостью к активированному белку C. Кровь. 1998;91:1140–1144. doi: 10.1182/кровь.V91.4.1140. [PubMed] [CrossRef] [Google Scholar]
74. Чжэн С., Чжан Б.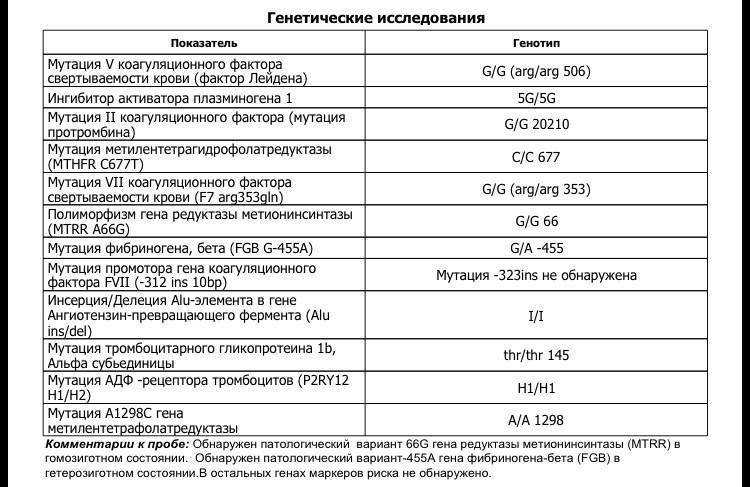 Комбинированный дефицит факторов свертывания крови V и VIII: обновление. Семин. тромб. Хемост. 2013; 39: 613–620. doi: 10.1055/s-0033-1349223. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
Комбинированный дефицит факторов свертывания крови V и VIII: обновление. Семин. тромб. Хемост. 2013; 39: 613–620. doi: 10.1055/s-0033-1349223. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
75. Эверетт Л.А., Хориати Р.Н., Чжан Б., Гинзбург Д. Измененный фенотип у мышей с дефицитом LMAN1 с низким уровнем остаточной экспрессии LMAN1. Кровь Adv. 2020;4:5635–5643. doi: 10.1182/bloodadvances.2020002523. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
76. Альтенхофф А.М., Гловер Н.М., Дессимос С. Вывод ортологии и паралогии. В: Анисимова М., редактор. Эволюционная геномика. Том. 1910. Спрингер; Нью-Йорк, штат Нью-Йорк, США: 2019. стр. 149–175. Методы молекулярной биологии. [PubMed] [CrossRef] [Google Scholar]
77. Ян Т.Л., Цуй Дж., Рехумтулла А., Ян А., Муссалли М., Кауфман Р.Дж., Гинзбург Д. Структура и функция мышиного фактора V и его инактивация по белку С. Кровь. 1998;91:4593–4599. doi: 10.1182/кровь.V91.12.4593. [PubMed] [CrossRef] [Google Scholar]
78. Rallapalli P.M., Orengo C.A., Studer R.A., Perkins S.J. Положительный отбор при эволюции факторов свертывания крови в контексте их болезнетворных мутаций. Мол. биол. Эвол. 2014;31:3040–3056. doi: 10.1093/molbev/msu248. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
Rallapalli P.M., Orengo C.A., Studer R.A., Perkins S.J. Положительный отбор при эволюции факторов свертывания крови в контексте их болезнетворных мутаций. Мол. биол. Эвол. 2014;31:3040–3056. doi: 10.1093/molbev/msu248. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
79. Mariz J.P.V., Nery M.F. Раскрытие молекулярной эволюции генов свертывания крови у рыб и китообразных. Передний. мар. 2020;7:592383. doi: 10.3389/fmars.2020.592383. [CrossRef] [Google Scholar]
80. Kretz C.A., Weyand A.C., Shavit J.A. Моделирование нарушений свертывания крови у рыбок данио. Курс. патобиол. Отчет 2015; 3: 155–161. doi: 10.1007/s40139-015-0081-3. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
81. Fish R.J., Freire C., Di Sanza C., Neerman-Arbez M. Венозный тромбоз и активность тромбоцитов в моделях количественных и качественных нарушений фибриногена у рыбок данио. . Междунар. Дж. Мол. науч. 2021;22:655. дои: 10.3390/ijms22020655. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
82. Iyer N., Jagadeeswaran P. Микрокинетические анализы коагуляции для плазмы человека и рыбок данио. Кровь. Коагул. Фибринолиз. 2021; 32: 50–56. doi: 10.1097/MBC.0000000000000975. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
Iyer N., Jagadeeswaran P. Микрокинетические анализы коагуляции для плазмы человека и рыбок данио. Кровь. Коагул. Фибринолиз. 2021; 32: 50–56. doi: 10.1097/MBC.0000000000000975. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
83. Kuta P., Melling N., Zimmermann R., Achenbach S., Eckstein R., Strobel J. Активность фактора свертывания крови в свежезамороженной плазме после Оттаивание с помощью нового устройства для оттаивания радиоволн. Переливание. 2019;59:1857–1861. doi: 10.1111/trf.15246. [PubMed] [CrossRef] [Google Scholar]
84. Марчесини Э., Морфини М., Валентино Л. Последние достижения в лечении гемофилии: обзор. БТТ. 2021; 15: 221–235. doi: 10.2147/BTT.S252580. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
85. Санкар А.Д., Вейанд А.С., Пайп С.В. Эволюция замены рекомбинантных факторов гемофилии. Трансфус. Афер. науч. 2019; 58: 596–600. doi: 10.1016/j.transci.2019.08.010. [PubMed] [CrossRef] [Академия Google]
86. Хай К.А. , Ронкароло М.Г. Генная терапия. Н. англ. Дж. Мед. 2019; 381: 455–464. doi: 10.1056/NEJMra1706910. [PubMed] [CrossRef] [Google Scholar]
, Ронкароло М.Г. Генная терапия. Н. англ. Дж. Мед. 2019; 381: 455–464. doi: 10.1056/NEJMra1706910. [PubMed] [CrossRef] [Google Scholar]
87. Anguela X.M., High K.A. Вступление в современную эру генной терапии. Анну. преподобный мед. 2019;70:273–288. doi: 10.1146/annurev-med-012017-043332. [PubMed] [CrossRef] [Google Scholar]
88. Камияма Ю., Наритоми Ю., Мория Ю., Ямамото С., Китахаши Т., Маэкава Т., Яхата М., Ханада Т., Утияма А., Ноумару А. и др. Исследования биораспределения продуктов клеточной терапии: текущее состояние и проблемы. Реген. тер. 2021; 18: 202–216. doi: 10.1016/j.reth.2021.06.005. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
89. Бужор Э., Лешанский Л., Блюменталь Дж., Бараш Х., Варшавский Д., Мазор Ю., Штричман Р. Подходы клеточной терапии: надежда на неизлечимые болезни. Реген. Мед. 2014; 9: 649–672. doi: 10.2217/rme.14.35. [PubMed] [CrossRef] [Google Scholar]
90. Weinshilboum R.M., Wang L. Pharmacogenomics: Precision Medicine and Drug Response.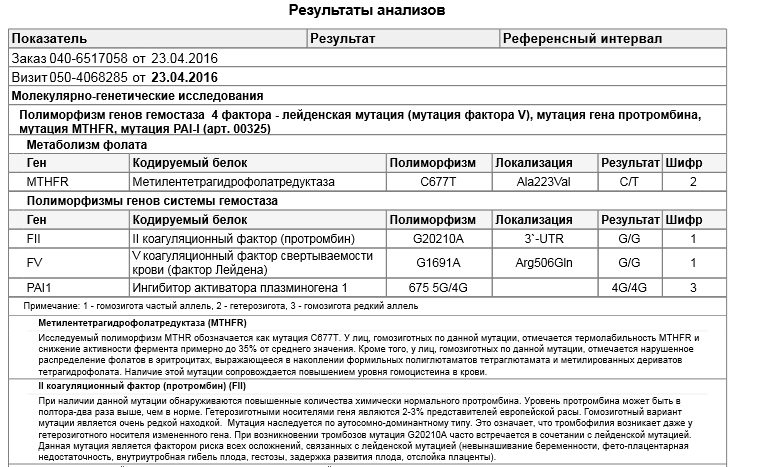 Мэйо Клин. проц. 2017;92:1711–1722. doi: 10.1016/j.mayocp.2017.09.001. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
Мэйо Клин. проц. 2017;92:1711–1722. doi: 10.1016/j.mayocp.2017.09.001. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
91. De Haan P., Van Diemen F.R., Toscano M.G. Векторы доставки вирусных генов: лекарства нового поколения для лечения заболеваний, связанных с иммунитетом. Гум. Вакцина. Иммунотер. 2021; 17:14–21. дои: 10.1080/21645515.2020.1757989. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
92. Athanasopoulos T., Munye M.M., Yáñez-Muñoz R.J. Неинтегрирующие векторы генной терапии. Гематол. Онкол. клин. Север Ам. 2017; 31: 753–770. doi: 10.1016/j.hoc.2017.06.007. [PubMed] [CrossRef] [Google Scholar]
93. Зу Х., Гао Д. Невирусные векторы в генной терапии: последние разработки, проблемы и перспективы. AAPS J. 2021; 23:78. doi: 10.1208/s12248-021-00608-7. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
94. Гевилер Э.К.Н., Мотта С.Е., Мартин М., Нуграха Б., Хёрструп С.П., Эммерт М.Ю. ИПСК человека и технологии редактирования генома для точной инженерии сердечно-сосудистой ткани. Передний. Сотовый Дев. биол. 2021;9:639699. doi: 10.3389/fcell.2021.639699. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
Передний. Сотовый Дев. биол. 2021;9:639699. doi: 10.3389/fcell.2021.639699. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
95. Гарсия-Бернал Д., Гарсия-Арранс М., Яньес Р.М., Эрвас-Сальседо Р., Кортес А., Фернандес-Гарсия М., Эрнандо-Родригес М., Кинтана-Бустаманте О., Бюрен Х.А., Гарсия-Олмо Д. и др. Текущее состояние мезенхимальных стромальных клеток: противоречия, нерешенные вопросы и некоторые многообещающие решения для повышения их терапевтической эффективности. Передний. Сотовый Дев. биол. 2021;9:650664. doi: 10.3389/fcell.2021.650664. [бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
96. Фомин М.Е., Тогаррати П.П., Мюнч М.О. Прогресс и проблемы в разработке клеточной терапии гемофилии А. Дж. Тромб. Гемост. 2014; 12:1954–1965. doi: 10.1111/jth.12750. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
97. Родригес-Мерчан Э.К., Де Пабло-Морено Дж.А., Лирас А. Генная терапия гемофилии: последние достижения. Междунар. Дж. Мол.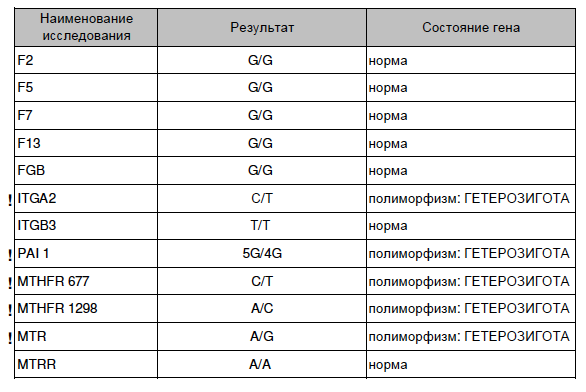 науч. 2021;22:7647. дои: 10.3390/ijms22147647. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
науч. 2021;22:7647. дои: 10.3390/ijms22147647. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
98. Парри М.С., Джанетти Дж., Душпанова А., Делла Пина Ф., Сарачини К., Маркуччи Р., Джусти Б., Берти С. Пантопразол значительно снижает антиагрегантный эффект клопидогреля: результаты пилотного рандомизированного исследования. Междунар. Дж. Кардиол. 2013;167:2177–2181. doi: 10.1016/j.ijcard.2012.05.080. [PubMed] [CrossRef] [Google Scholar]
99. Триподи А., Арбини А., Чантарангкул В., Маннуччи П.М. Рекомбинантный тканевый фактор как замена обычному тромбопластину в тесте протромбинового времени. тромб. Гемост. 1992;67:42–45. doi: 10.1055/s-0038-1648376. [PubMed] [CrossRef] [Google Scholar]
100. Van den Besselaar A.M., Neuteboom J., Bertina R.M. Влияние синтетических фосфолипидов на реакцию активированного частичного тромбопластинового времени на гепарин. Кровавый коагул. Фибринолиз. 1993; 4: 895–903. doi: 10.1097/00001721-199312000-00006. [PubMed] [CrossRef] [Google Scholar]
101. Ray M.J., Hawson G.A.T. Сравнение двух реагентов АЧТВ, в которых используются активаторы кремнезема. клин. лаборатория Гематол. 1989;11:221–232. doi: 10.1111/j.1365-2257.1989.tb00212.x. [PubMed] [CrossRef] [Google Scholar]
Ray M.J., Hawson G.A.T. Сравнение двух реагентов АЧТВ, в которых используются активаторы кремнезема. клин. лаборатория Гематол. 1989;11:221–232. doi: 10.1111/j.1365-2257.1989.tb00212.x. [PubMed] [CrossRef] [Google Scholar]
102. Den Dunnen J.T., Dalgleish R., Maglott D.R., Hart R.K., Greenblatt M.S., McGowan-Jordan J., Roux A.F., Smith T., Antonarakis S.E., Taschner P.E.M., и другие. Рекомендации HGVS по описанию вариантов последовательности: обновление 2016 г. Гум. Мутат. 2016; 37: 564–569. doi: 10.1002/humu.22981. [PubMed] [CrossRef] [Google Scholar]
103. Di Tommaso P., Moretti S., Xenarios I., Orobitg M., Montanyola A., Chang J.M., Taly J.F., Notredame C. T-Coffee: A Web Сервер для множественного выравнивания последовательностей белков и последовательностей РНК с использованием структурной информации и расширения гомологии. Нуклеиновые Кислоты Res. 2011;39:В13–В17. doi: 10.1093/nar/gkr245. [Бесплатная статья PMC] [PubMed] [CrossRef] [Google Scholar]
Ген F9: MedlinePlus Genetics
фактор свертывания крови IX
Чтобы использовать функции обмена на этой странице, включите JavaScript.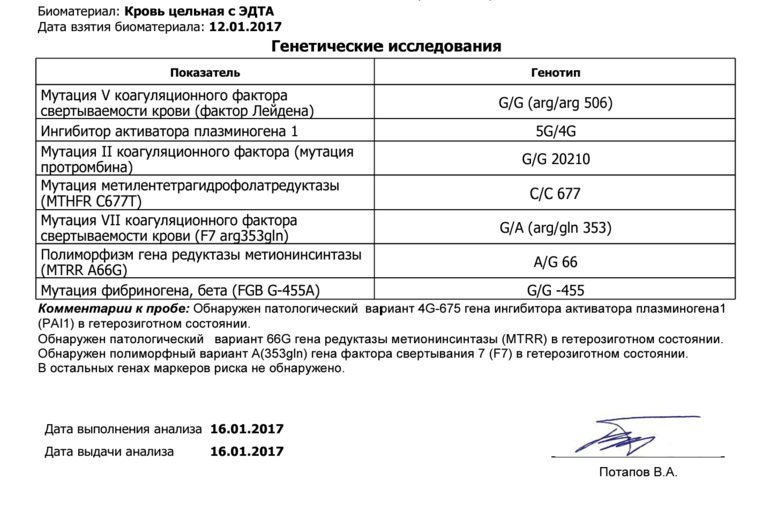
Нормальное функционирование
Ген F9 предоставляет инструкции для производства белка, называемого фактором свертывания крови IX. Факторы свертывания крови представляют собой группу родственных белков, которые необходимы для образования тромбов. После травмы сгустки защищают тело, закупоривая поврежденные кровеносные сосуды и предотвращая дальнейшую потерю крови.
Фактор свертывания крови IX вырабатывается в печени. Этот белок циркулирует в кровотоке в неактивной форме до тех пор, пока не произойдет травма, повреждающая кровеносные сосуды. В ответ на повреждение фактор свертывания крови IX активируется другим фактором свертывания крови, называемым фактором XIa. Активный белок (иногда обозначаемый как фактор свертывания крови IXa) взаимодействует с фактором свертывания крови VIII и другими молекулами. Эти взаимодействия запускают цепочку дополнительных химических реакций, в результате которых образуется тромб.
Заболевания, связанные с генетическими изменениями
Гемофилия
Мутации в гене F9 вызывают тип гемофилии, называемый гемофилией В.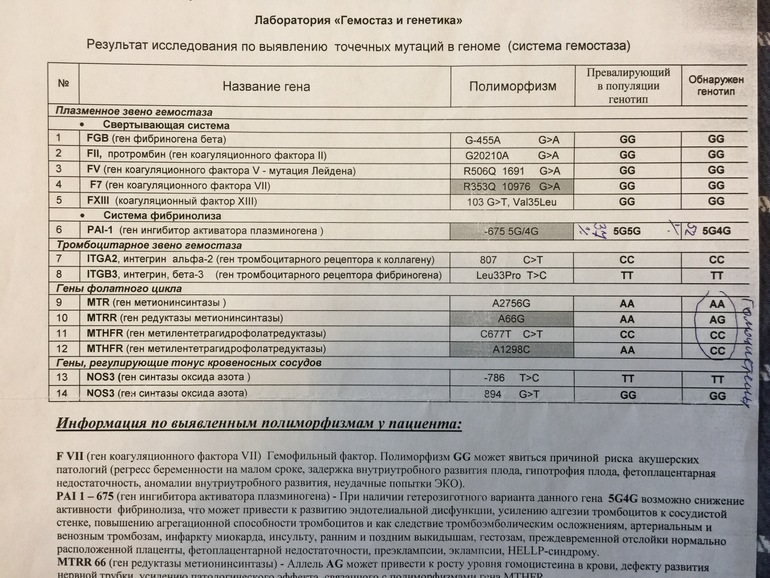 Было идентифицировано более 900 изменений в этом гене. Наиболее распространенные мутации изменяют отдельные строительные блоки ДНК (пары оснований) в гене. Небольшой процент мутаций удаляет или вставляет несколько пар оснований или перестраивает сегменты ДНК внутри гена.
Было идентифицировано более 900 изменений в этом гене. Наиболее распространенные мутации изменяют отдельные строительные блоки ДНК (пары оснований) в гене. Небольшой процент мутаций удаляет или вставляет несколько пар оснований или перестраивает сегменты ДНК внутри гена.
Мутации в гене F9 приводят к выработке аномальной версии фактора свертывания IX или снижению количества этого белка. Измененный или отсутствующий белок не может эффективно участвовать в процессе свертывания крови. В результате сгустки крови не могут образовываться должным образом в ответ на травму. Эти проблемы со свертываемостью крови приводят к чрезмерному кровотечению, которое бывает трудно контролировать. Мутации, полностью устраняющие активность фактора свертывания IX, приводят к тяжелой гемофилии. Мутации, снижающие, но не устраняющие активность белка, обычно вызывают легкую или умеренную гемофилию.
Несколько мутаций в начале последовательности гена F9 вызывают необычную форму гемофилии, известную как гемофилия B Лейдена.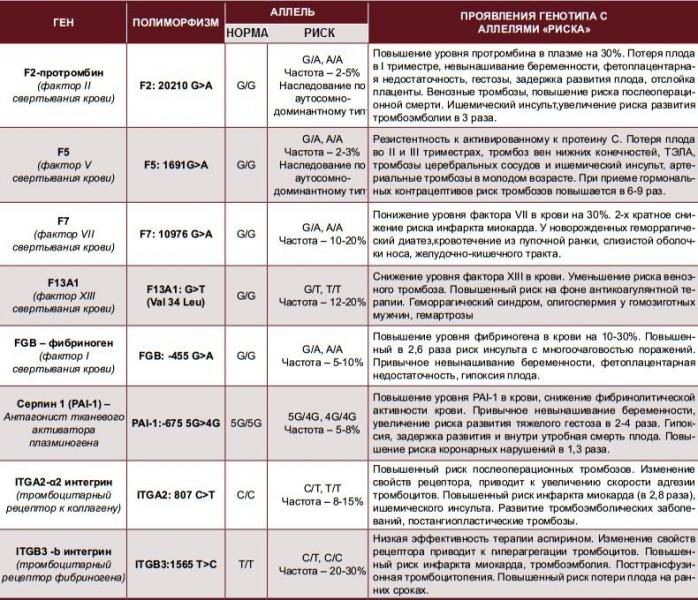 Люди с этими мутациями рождаются с очень низким уровнем функционального фактора свертывания крови IX, но гормональные изменения вызывают постепенное повышение уровня этого белка в период полового созревания. В результате у взрослых с гемофилией B Leyden редко возникают эпизоды аномального кровотечения.
Люди с этими мутациями рождаются с очень низким уровнем функционального фактора свертывания крови IX, но гормональные изменения вызывают постепенное повышение уровня этого белка в период полового созревания. В результате у взрослых с гемофилией B Leyden редко возникают эпизоды аномального кровотечения.
Подробнее об этом заболевании
Чувствительность к варфарину
MedlinePlus Genetics предоставляет информацию о чувствительности к варфарину
Подробнее об этом заболевании
Другие заболевания
Несколько редких мутаций в гене F9 вызывают повышенную чувствительность (гиперчувствительность) к препарату под названием варфарин. Это лекарство является антикоагулянтом, что означает, что оно используется для предотвращения образования или роста аномальных тромбов. Варфарин работает, уменьшая количество активного фактора IX и трех других белков свертывания крови.
Каждая из мутаций, ответственных за гиперчувствительность к варфарину, изменяет одну пару оснований в гене F9 . Эти мутации не вызывают гемофилию В, и у людей с такими генетическими изменениями проблемы с кровотечением возникают только в том случае, если их лечат варфарином. Варфарин снижает количество фактора свертывания крови IX до очень низкого уровня у этих людей, что препятствует нормальному свертыванию крови и может привести к повторяющимся тяжелым проблемам с кровотечением. Чтобы избежать этих осложнений, людей с гиперчувствительностью к варфарину можно лечить другими антикоагулянтами.
Эти мутации не вызывают гемофилию В, и у людей с такими генетическими изменениями проблемы с кровотечением возникают только в том случае, если их лечат варфарином. Варфарин снижает количество фактора свертывания крови IX до очень низкого уровня у этих людей, что препятствует нормальному свертыванию крови и может привести к повторяющимся тяжелым проблемам с кровотечением. Чтобы избежать этих осложнений, людей с гиперчувствительностью к варфарину можно лечить другими антикоагулянтами.
Other Names for This Gene
- Christmas factor
- coagulation factor IX (plasma thromboplastic component, Christmas disease, hemophilia B)
- FA9_HUMAN
- Factor 9
- FIX
- HEMB
- Plasma thromboplastin component
- PTC
Дополнительная информация и ресурсы
Тесты, внесенные в Реестр генетических тестов
- Тесты F9
Научные статьи в PubMed
- PubMed
Каталог генов и болезней от OMIM
- ФАКТОР КОАГУЛЯЦИИ IX
Базы данных генов и вариантов
- Ген NCBI
- КлинВар
Ссылки
- Bolton-Maggs PH, Pasi KJ. Гемофилии А и В. Ланцет. 2003 май
24;361(9371):1801-9. doi: 10.1016/S0140-6736(03)13405-8. Цитата в PubMed
- Боуэн Диджей. Гемофилия А и гемофилия В: молекулярные идеи. Мол Патол. 2002 г., апрель; 55 (2): 127–44. doi: 10.1136/mp.55.2.127. Опечатка в: Мол Патол 2002 Июнь; 55 (3): 208. Цитирование в PubMed или бесплатная статья в PubMed Central
- Чу К., Ву С.М., Стэнли Т., Стаффорд Д.В., Хай К.А. Мутация в пропептиде Фактор IX приводит к чувствительности к варфарину по новому механизму. Джей Клин Инвест. 1996 г. 1 октября 98(7):1619-25. DOI: 10.1172/JCI118956. Цитирование в PubMed или бесплатная статья в PubMed Central
- Джангранде П. Гемофилия B: Рождественская болезнь. Эксперт Опин Фармаколог. 2005 г. 6(9) августа: 1517-24. дои: 10.1517/14656566.6.9.1517. Цитата в PubMed
- Кристенсен С.Р. Лечение варфарином пациента с фактором свертывания крови IX
Мутация пропептида вызывает гиперчувствительность к варфарину. Кровь. 2002 Октябрь
1;100(7):2676-7. doi: 10.1182/кровь-2002-06-1753. Аннотация недоступна. Цитата в PubMed
- Лилликрап D. Молекулярные основы гемофилии B. Гемофилия. 1998 Июль; 4 (4): 350-7. doi: 10.1046/j.1365-2516.1998.440350.x. Цитата в PubMed
- Олденбург Дж., Криз К., Вуйлемин В.А., Малый Ф.Е., фон Фельтен А., Зигемунд А., Килинг Д.М., Бейкер П., Чу К., Конкл Б.А., Ламмле Б., Альберт Т.; Исследовательская группа по наследственным Чувствительность к варфарину. Генетическая предрасположенность к кровотечениям при оральном антикоагулянтная терапия: доказательства распространенных мутаций-основателей (FIXVal-10 и FIXThr-10) и независимая мутация горячей точки CpG (FIXThr-10). Тромб Хемост. 2001 март; 85 (3): 454-7. Цитата на PubMed
- Ольденбург Дж., Квенцель Э.М., Харбрехт Ю., Фрегин А., Кресс В., Мюллер К.Р., Хертфельдер
HJ, Schwaab R, Brackmann HH, Hanfland P. Мутации Missense в ALA-10 в
пропептид фактора IX: незначительный вариант в нормальной жизни, но решающий
причиной кровотечения во время пероральной антикоагулянтной терапии.
 Гемофилии А и В. Ланцет. 2003 май
24;361(9371):1801-9. doi: 10.1016/S0140-6736(03)13405-8. Цитата в PubMed
Гемофилии А и В. Ланцет. 2003 май
24;361(9371):1801-9. doi: 10.1016/S0140-6736(03)13405-8. Цитата в PubMed Кровь. 2002 Октябрь
1;100(7):2676-7. doi: 10.1182/кровь-2002-06-1753. Аннотация недоступна. Цитата в PubMed
Кровь. 2002 Октябрь
1;100(7):2676-7. doi: 10.1182/кровь-2002-06-1753. Аннотация недоступна. Цитата в PubMed