
Википедия хорея гентингтона: HTTP status 402 — payment required, требуется оплата
About Huntington’s Disease — European Huntington’s Disease Network
БГ названа в честь Джорджа Гентингтона, американского врача, подробно описавшего это заболевание в 1872 г. Его описание основано на наблюдениях семей с БГ из деревни Ист-Хэмптон, расположенной на острове Лонг-Айленд (Нью-Йорк, США), где жил и работал д-р Гентингтон. БГ известна в прошлом как хорея Гентингтона и пляска святого Витта.
БГ является редким заболеванием, распространённость которого в европейской популяции составляет около 5–10 человек на 100 000 населения. Примерно такая же распространённость и в странах, население которых имеет преимущественно европейское происхождение, например, в США. Распространённость БГ меньше в странах Азии и Африки и составляет около 1 человека на 100 000 населения. Мужчины и женщины имеют одинаковые риски унаследовать экспансию, которая приведёт к развитию БГ. БГ характеризуется сочетанием двигательных (моторных), поведенческих (например, изменение настроения) и когнитивных (например, трудности в понимании) нарушений, однако могут развиваться и другие симптомы. От пациента к пациенту (даже среди членов одной и той же семьи) симптомы БГ могут отличаться по своей тяжести, возрасту появления и темпу прогрессирования. У одного человека могут быть выраженные двигательные расстройства, но при этом лёгкие поведенческие и когнитивные нарушения, в то время как у другого депрессия и тревога могут наблюдаться за годы до каких-либо двигательных симптомов. Про БГ пишут, что она развивается исподволь, т.к. довольно сложно определить точную дату появления её первых симптомов. У большинства людей, являющихся носителями мутации БГ, симптомы заболевания развиваются в среднем возрасте — между 35 и 55 годами жизни.Как часто встречается БГ?

Каковы симптомы БГ?
К двигательным симптомам БГ относятся хорея, брадикинезия и дистония, которые в значительной степени нарушают поддержание физиологической позы, равновесие и ходьбу.
К наиболее частым психопатологическим симптомам при БГ относятся апатия, тревога, депрессия, раздражительность, вспышки агрессии, импульсивное поведение, обсессивно-компульсивные нарушения, нарушение сна и социальное отчуждение. Реже встречаются мания и шизофреноподобные проявления, включая бред (ложные убеждения) и галлюцинации (зрительное, слуховое или иное восприятие вещей, не существующих на самом деле). У лиц с БГ могут также отмечаться суицидальные мысли (особенно на ранних стадиях заболевания). Большинство пациентов и ухаживающих лиц считают поведенческие изменения более обременительными, чем двигательные или когнитивные нарушения вследствие БГ.
У лиц с БГ могут также отмечаться суицидальные мысли (особенно на ранних стадиях заболевания). Большинство пациентов и ухаживающих лиц считают поведенческие изменения более обременительными, чем двигательные или когнитивные нарушения вследствие БГ.
БГ характеризуется постепенным нарушением способности понимания и логического суждения, а также снижением памяти. Когнитивные нарушения включают в себя замедление мышления и затруднение концентрации внимания, сложности в организации, планировании, принятии решений и в ответах на вопросы, снижение краткосрочной памяти в сочетании с затруднениями в решении проблем, понимании и усвоении новой информации.
По мере течения БГ у человека могут наблюдаться и другие изменения, такие как снижение аппетита, потеря массы тела, снижение самооценки, нарушение сексуального влечения, а также недержание мочи и стула.Когда появляются симптомы БГ?
 Примерно в 10 % случаев болезнь дебютирует до 20 лет (в таком случае имеет место ювенильная БГ), а в ещё 10 % — после 55 лет. В целом, БГ развивается постепенно, в связи с чем она может оставаться недиагностированной в течение многих лет. В среднем, продолжительность жизни от момента постановки диагноза составляет от 15 до 20 лет, но эти рамки значительно варьируют среди пациентов и могут сильно зависеть от качества ухода за больным.
Примерно в 10 % случаев болезнь дебютирует до 20 лет (в таком случае имеет место ювенильная БГ), а в ещё 10 % — после 55 лет. В целом, БГ развивается постепенно, в связи с чем она может оставаться недиагностированной в течение многих лет. В среднем, продолжительность жизни от момента постановки диагноза составляет от 15 до 20 лет, но эти рамки значительно варьируют среди пациентов и могут сильно зависеть от качества ухода за больным.
Факторы, определяющие возраст начала заболевания, довольно сложны и являются предметом проводимых в настоящее время исследований. При анализе больших групп пациентов с БГ учёные выявили корреляцию между числом тринуклеотидных повторов и возрастом появления симптомов (см. рисунок ниже). Это означает, что, в целом, чем больше CAG-повторов, тем раньше появляются симптомы (см. рисунок ниже).От чего зависит возраст появления симптомов?
 Тем не менее, как видно на рисунке ниже, для каждого конкретного числа СAG-повторов предполагаемый возраст дебюта заболевания может варьировать в пределах 30 лет. Возможно, такая вариабельность связана с действием других генов, отличных от
Тем не менее, как видно на рисунке ниже, для каждого конкретного числа СAG-повторов предполагаемый возраст дебюта заболевания может варьировать в пределах 30 лет. Возможно, такая вариабельность связана с действием других генов, отличных от  Рисунок адаптирован из Andrew, S. E. et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nature Genetics 4, 398–403 (1993).
Рисунок адаптирован из Andrew, S. E. et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nature Genetics 4, 398–403 (1993).
Согласно классификации, разработанной неврологом и специалистом по БГ Айрой Шолсеном (Ira Shoulson) из Джорджтаунского университета (США), течение БГ можно разделить на пять стадий.Какие стадии выделяют в течении БГ?

Если БГ начинается в раннем возрасте (до 20 лет), наиболее выраженными симптомами являются замедленность движений (брадикинезия) и скованность (дистония), а не насильственные движения (хорея). К ранним симптомам ювенильной БГ относятся поведенческие нарушения, трудности в обучении и речи, снижение успеваемости в школе. В ряде случаев могут наблюдаться эпилептические приступы, что характерно для пациентов более молодого возраста.Отличаются ли симптомы ювенильной формы БГ от симптомов
заболевания с началом во взрослом возрасте?  В целом, ювенильная форма БГ прогрессирует быстрее, чем БГ с дебютом во взрослом возрасте.
В целом, ювенильная форма БГ прогрессирует быстрее, чем БГ с дебютом во взрослом возрасте.
Если БГ начинается в позднем возрасте, наиболее выраженным симптомом является хорея, а не замедленность или скованность движений. В таких случаях нередко сложно выявить семейный характер заболевания, т.к. родители пациента, чаще всего, уже скончались, по всей видимости, не дожив до того возраста, когда бы у них развились симптомы БГ.Каковы симптомы БГ, если заболевание развивается в
позднем возрасте?
Люди с БГ не умирают непосредственно от этого заболевания — причиной смерти являются другие медицинские состояния, являющиеся осложнениями общего истощения организма. Последние включают в себя пневмонию (она является причиной смерти при БГ в трети случаев), попёрхивание, сердечную недостаточность, травмы головы вследствие падений и нарушение питания.Причины смерти
 Высок также и риск суицида — он является причиной смерти примерно у 7 % пациентов.
Высок также и риск суицида — он является причиной смерти примерно у 7 % пациентов.
Диагноз БГ ставится на основании сочетания результатов клинического обследования и генетического теста. Клинический диагноз базируется на данных истории болезни и семейного анамнеза, а также на данных стандартного обследования с использованием клинических шкал, которые оценивают частоту и выраженность симптомов БГ. Клинический диагноз, как правило, подтверждается проведением генетического тестирования на наличие мутации (экспансии) в гене HTT (так называемое диагностическое, или подтверждающее, генетическое тестирование). Если у человека нет никаких симптомов, но он находится в группе риска по носительству мутации БГ, определить его генетический статус может пресимптоматическое генетическое тестирование (известное также как предиктивное генетическое тестирование). Каким образом ставится диагноз БГ?

Методы клинического обследования, которые используются для диагностики БГ и оценки различных её проявлений, не являются одинаковыми во всех больницах и во всех странах. Тем не менее, наиболее часто используемой является Унифицированная шкала оценки болезни Гентингтона (англ. Unified Huntington’s Disease Rating Scale, UHDRS), имеющая подразделы для двигательной, когнитивной оценки, а также оценки поведенческих и функциональных нарушений. Кроме того, часто используется Шкала оценки поведенческих нарушений при болезни Гентингтона (англ. Problem Behaviours Assessment for Huntington’s Disease, PBA) для исследования выраженности и частоты нарушений поведения (таких как сниженное настроение, апатия и раздражительность) и ряд других тестов. Например, Краткая шкала оценки психического статуса (англ.Какие методы клинического обследования используются для
диагностиrи БГ? Mini-Mental State Examination, MMSE) и Шкала Маттиса для оценки деменции (англ. Mattis Dementia Rating Scale) применяются дополнительно к подразделу UHDRS по оценке когнитивных нарушений.
Mini-Mental State Examination, MMSE) и Шкала Маттиса для оценки деменции (англ. Mattis Dementia Rating Scale) применяются дополнительно к подразделу UHDRS по оценке когнитивных нарушений.
Знание того, что у вас есть риск заболеть БГ, может привести к чувству постоянного беспокойства в течение жизни. По этой причине человек может посчитать, что лучше знать наверняка, есть ли у него мутация этого заболевания или нет. В таком случае крайне рекомендуется проведение генетического консультирования с психологической поддержкой, т.к. это позволяет лучше понять возможные пути для выбора и обсудить беспокоящие вопросы. В целом, пресимптоматическое тестирование не рекомендуется для лиц младше 18 лет — возраста, в котором, как считается, человек способен воспринять информацию о том, что он является носителем мутации БГ.Какова процедура прохождения пресимптоматического генетического тестирования?
 Тем не менее, в порядке исключения детям может проводиться подтверждающее генетическое тестирование, если, например, у ребёнка имеются симптомы ювенильной формы БГ или если женщина моложе 18 лет забеременела. Если вы решили пройти генетическое тестирование, из вены на вашей руке возьмут образец крови, чтобы извлечь из неё молекулы ДНК. В зависимости от условий лаборатории, где сдавалась кровь, результаты будут готовы через 2–8 недель. В 2012 г. рабочая группа EHDN по генетическому тестированию и консультированию обновила
Тем не менее, в порядке исключения детям может проводиться подтверждающее генетическое тестирование, если, например, у ребёнка имеются симптомы ювенильной формы БГ или если женщина моложе 18 лет забеременела. Если вы решили пройти генетическое тестирование, из вены на вашей руке возьмут образец крови, чтобы извлечь из неё молекулы ДНК. В зависимости от условий лаборатории, где сдавалась кровь, результаты будут готовы через 2–8 недель. В 2012 г. рабочая группа EHDN по генетическому тестированию и консультированию обновила
Рекомендации по процедуре пресимптоматического генетического тестирования.
Генетическое тестирование определяет число CAG-повторов в гене HTT. Тест позволяет узнать, есть ли у человека мутация БГ или нет, однако он не даёт информации о возрасте дебюта этого заболевания, скорости его прогрессирования или о том, какие именно симптомы разовьются.Что определяет генетическое тестирование?
 Точность результата генетического тестирования близка к 100 %. Результаты анализа ДНК, как правило, дважды перепроверяются с использованием двух разных образцов крови. Кроме того, для подтверждения исходного диагноза может быть также взята на анализ кровь родителя обследуемого лица (или, в случае отсутствия такой возможности, другого члена семьи).
Точность результата генетического тестирования близка к 100 %. Результаты анализа ДНК, как правило, дважды перепроверяются с использованием двух разных образцов крови. Кроме того, для подтверждения исходного диагноза может быть также взята на анализ кровь родителя обследуемого лица (или, в случае отсутствия такой возможности, другого члена семьи).
Да. Это можно сделать с использованием современной диагностической процедуры под названием преимплантационная генетическая диагностика (ПГД), или скрининг эмбрионов, которая применяется в сочетании с экстракорпоральным оплодотворением (ЭКО) и включает в себя скрининг эмбрионов на носительство мутации БГ до их имплантации в матку. Использование этой технологии позволяет быть уверенным в том, что имплантированы будут только эмбрионы с нормальным числом CAG-повторов.Возможно ли человеку, являющемуся носителем мутации БГ,
предотвратить передачу этой мутации своему ребёнку? Таким образом, даже если у кого-то из родителей есть мутация БГ (вне зависимости от того, мужчина это или женщина), ПГД позволяет паре зачать ребёнка, не являющегося носителем мутации БГ. Тем не менее, в некоторых странах приняты законы о защите эмбриона, не позволяющие проводить ПГД. Также необходимо знать, что шансы на успешное завершение беременности после ПГД/ЭКО ниже, чем при «естественном» зачатии. В некоторых странах возможно тестирование нерождённого плода, зачатого естественным образом, с целью дальнейшего решения о возможном прерывании беременности на основании знания генетического статуса плода.
Таким образом, даже если у кого-то из родителей есть мутация БГ (вне зависимости от того, мужчина это или женщина), ПГД позволяет паре зачать ребёнка, не являющегося носителем мутации БГ. Тем не менее, в некоторых странах приняты законы о защите эмбриона, не позволяющие проводить ПГД. Также необходимо знать, что шансы на успешное завершение беременности после ПГД/ЭКО ниже, чем при «естественном» зачатии. В некоторых странах возможно тестирование нерождённого плода, зачатого естественным образом, с целью дальнейшего решения о возможном прерывании беременности на основании знания генетического статуса плода.
Пренатальная (до рождения) диагностика возможна только в том случае, когда запрашивающие её люди могут показать, что их случай удовлетворяет определённым медицинским и юридическим критериям, которые варьируют в зависимости от страны.Могу ли я провести генетическое тестирование своему ребёнку,
который ещё не родился? Существует две стандартные процедуры проведения пренатальной диагностики. Первая — это амниоцентез (известна также как исследование амниотической жидкости), при котором при помощи вводимой через стенку живота матери иглы собирается амниотическая жидкость, содержащая клетки плода. Как правило, амниоцентез проводится после 14-й недели беременности. Вторая процедура для пренатальной диагностики — биопсия ворсин хориона, при которой забирается образец ворсины хориона (плацентарная ткань). Исследование ворсин хориона может проводиться раньше — между 10-й и 13-й неделями беременности, однако это более опасная для плода процедура.
Существует две стандартные процедуры проведения пренатальной диагностики. Первая — это амниоцентез (известна также как исследование амниотической жидкости), при котором при помощи вводимой через стенку живота матери иглы собирается амниотическая жидкость, содержащая клетки плода. Как правило, амниоцентез проводится после 14-й недели беременности. Вторая процедура для пренатальной диагностики — биопсия ворсин хориона, при которой забирается образец ворсины хориона (плацентарная ткань). Исследование ворсин хориона может проводиться раньше — между 10-й и 13-й неделями беременности, однако это более опасная для плода процедура.
На текущий момент не существует методов, позволяющих эффективно воздействовать на причину развития БГ. Тем не менее, в течение последних лет фундаментальные и клинические исследования значительно расширили наши знания о БГ, и , направленных на изучение патогенеза этого заболевания, с целью разработки препаратов, которые смогут отсрочить начало болезни или замедлить её прогрессирование.Есть ли какие-либо методы лечения БГ?
 На сегодняшний день имеются подходы, применение которых уменьшает выраженность симптомов БГ (симптоматическое лечение), что позволяет улучшить качество жизни пациентов. Эти подходы подразделяются на медикаментозное (лекарственное) и немедикаментозное (нелекарственное) лечение.
На сегодняшний день имеются подходы, применение которых уменьшает выраженность симптомов БГ (симптоматическое лечение), что позволяет улучшить качество жизни пациентов. Эти подходы подразделяются на медикаментозное (лекарственное) и немедикаментозное (нелекарственное) лечение.
Хорея, брадикинезия, раздражительность, апатия, депрессия, тревога и нарушение сна являются теми симптомами БГ, которые причиняют наибольшие трудности. Существует несколько вариантов медикаментозной коррекции этих симптомов. Тем не менее, многие лекарства могут вызывать нежелательные реакции или уменьшать эффективность других сопутствующих препаратов. Кроме того, одно и то же лекарство у разных людей может приводить к различным эффектам. Лечение должно проводиться и быть персонализированным, строящимся с учётом имеющихся у пациента симптомов и его/её ответа на пробуемые лекарства.Какие существуют методы медикаментозной коррекции симптомов БГ?
Немедикаментозные методы лечения (такие как психотерапия, когнитивная терапия, лечебная физкультура, логопедические занятия, дыхательная и трудотерапия) могут способствовать уменьшению выраженности как психопатологических, так и физических симптомов БГ. В частности, показано, что после применения перечисленных подходов отмечается улучшение настроения, контроля произвольных движений, речи, равновесия, глотания и ходьбы. Хорошо известно, что физические упражнения укрепляют и психическое (включая уменьшение симптомов депрессии), и физическое здоровье, способствуя улучшению общего самочувствия. Всё больше данных накапливается в пользу того, что физические занятия помогают также замедлить прогрессирование двигательных расстройств при БГ. Например, некоторые программы лечебной физкультуры показали свою эффективность в отношении контроля движения, ходьбы и равновесия.Как могут помочь немедикаментозные методы лечения?
 Рабочая группа EHDN по физиотерапии опубликовала для реабилитологов, работающих с пациентами с БГ.
Рабочая группа EHDN по физиотерапии опубликовала для реабилитологов, работающих с пациентами с БГ.
Потенциальная польза от диеты, обогащённой витаминами, коферментами и иными веществами являлась предметом большого числа обсуждений, однако пока положительное влиянияние такой диеты в отношении БГ так и не доказано клинически. Тем не менее, для некоторых пациентов с БГ, особенно на поздних стадиях заболевания, одной из проблем является снижение массы тела, в связи с чем очень важно обеспечить в течение болезни здоровое питание. На поздних стадиях может потребоваться высококалорийная диета. Также может быть оправданным направление на консультацию к диетологу.Может ли какая-то особая диета уменьшить выраженность
симптомов БГ?
БГ развивается вследствие изменения (увеличения — экспансии — копий одного из участков) гена (HTT), кодирующего белок под названием гентингтин.Что приводит к развитию БГ?
 Из-за этой экспансии с гена образуется изменённый вариант белка, что, в свою очередь, приводит к нарушению функционирования и гибели нервных клеток (нейронов) в определённых участках головного мозга. Поскольку у гентингтина имеется множество функций, точные механизмы развития заболевания сложны и многогранны. Для того, чтобы разработать методы лечения, изменяющие течение болезни, исследователи работают над подробным изучением механизмов её развития.
Из-за этой экспансии с гена образуется изменённый вариант белка, что, в свою очередь, приводит к нарушению функционирования и гибели нервных клеток (нейронов) в определённых участках головного мозга. Поскольку у гентингтина имеется множество функций, точные механизмы развития заболевания сложны и многогранны. Для того, чтобы разработать методы лечения, изменяющие течение болезни, исследователи работают над подробным изучением механизмов её развития.
В 1993 г. учёные выявили мутацию, приводящую к развитию БГ. Ген HTT расположен на 4-й хромосоме и кодирует белок под названием гентингтин. Этот ген содержит последовательность из трёх нуклеотидов (базовых компонентов ДНК) — цитозин-аденин-гуанин (CAG) — которая несколько раз повторяется. Число этих так называемых тринуклеотидных повторов может варьировать. Если у человека имеется 40 или более CAG-повторов в одной из копий гена HTT, то в течение жизни (при условии её нормальной продолжительности), т. Ген гентингтина (HTT) расположен на 4-й хромосоме. В нём содержатся повторяющиеся последовательности трёх базовых компонентов ДНК — C-A-G. Если у человека имеется 40 или более повторов C-A-G, то в течение жизни (при условии её нормальной продолжительности) у него разовьётся болезнь Гентингтона.Что является причиной развития БГ?
 е. в среднем возрасте, у него разовьётся БГ. Поскольку мутация, являющаяся причиной развития БГ, присутствует во всех клетках организма с самого зачатия и может передаваться последующим поколениям, это заболевание является наследственным.
е. в среднем возрасте, у него разовьётся БГ. Поскольку мутация, являющаяся причиной развития БГ, присутствует во всех клетках организма с самого зачатия и может передаваться последующим поколениям, это заболевание является наследственным.
По мере увеличения числа CAG-повторов определённый участок ДНК становится более нестабильным. Нестабильность означает, что при передаче следующему поколению число этих повторов может либо увеличиться, либо уменьшиться. Если число CAG-повторов в гене HTT менее 27, то этот участок стабилен.Что означает число CAG-повторов в гене
HTT? Если число повторов находится между 27 и 35 (так называемое промежуточное число повторов), у человека не разовьётся БГ, т.к. это укладывается в нормальные значения. Однако число CAG-повторов в 27 и более нестабильно, и существует риск увеличения их числа в следующем поколении, что обусловливает риск развития БГ у этих детей. У лиц, имеющих от 36 до 39 CAG-повторов, БГ может развиться, но только в пожилом возрасте. Этот диапазон известен как число повторов с неполной пенетрантностью. В случае наличия у человека более 39 CAG-повторов в течение жизни (при условии её нормальной продолжительности) у него разовьётся БГ — чаще всего, в среднем возрасте. В редких случаях число CAG-повторов может быть исключительно большим, что приводит к дебюту заболевания в подростковом или детском возрасте (ювенильная БГ). У пациентов с началом заболевания до 10 лет нередко имеется более 80 CAG-повторов.
Если число повторов находится между 27 и 35 (так называемое промежуточное число повторов), у человека не разовьётся БГ, т.к. это укладывается в нормальные значения. Однако число CAG-повторов в 27 и более нестабильно, и существует риск увеличения их числа в следующем поколении, что обусловливает риск развития БГ у этих детей. У лиц, имеющих от 36 до 39 CAG-повторов, БГ может развиться, но только в пожилом возрасте. Этот диапазон известен как число повторов с неполной пенетрантностью. В случае наличия у человека более 39 CAG-повторов в течение жизни (при условии её нормальной продолжительности) у него разовьётся БГ — чаще всего, в среднем возрасте. В редких случаях число CAG-повторов может быть исключительно большим, что приводит к дебюту заболевания в подростковом или детском возрасте (ювенильная БГ). У пациентов с началом заболевания до 10 лет нередко имеется более 80 CAG-повторов.Число CAG-повторов Разовьётся ли болезнь? Последствия для потомства Название состояния Менее 27 Нет Нет Нормальное число повторов 27–35 Нет Число CAG-повторов от 27 до 35 является нестабильным и может увеличиться при передаче следующему поколению Промежуточное число повторов 36–39 Вероятно Есть.  Для каждого из детей риск унаследовать мутацию составляет 50 %
Для каждого из детей риск унаследовать мутацию составляет 50 % Увеличение числа повторов с неполной пенетрантностью 40 и более Да Есть. Для каждого из детей риск унаследовать мутацию составляет 50 % Увеличение числа повторов с полной пенетрантностью
Гены находятся на хромосомах внутри каждой клетки нашего тела. Ген — это участок ДНК, содержащий код для образования определённого белка; с ДНК синтезируется матричная РНК (мРНК), с которой затем образуется белок. В большинстве случаев каждый человек наследует по две копии каждого гена: одну от мамы и одну от папы. При БГ поражается ген HTT, который кодирует белок под названием гентингтин. Если ребёнок наследует копию гена HTT с увеличенным числом CAG-повторов, у него тоже разовьётся БГ. Пояснение к рисунку. Гены располагаются вдоль хромосом, как бусинки на леске (изображены в виде разноцветных полос). В большинстве случаев каждый человек наследует по две копии каждого гена: одну от мамы и одну от папы.Что такое ген?
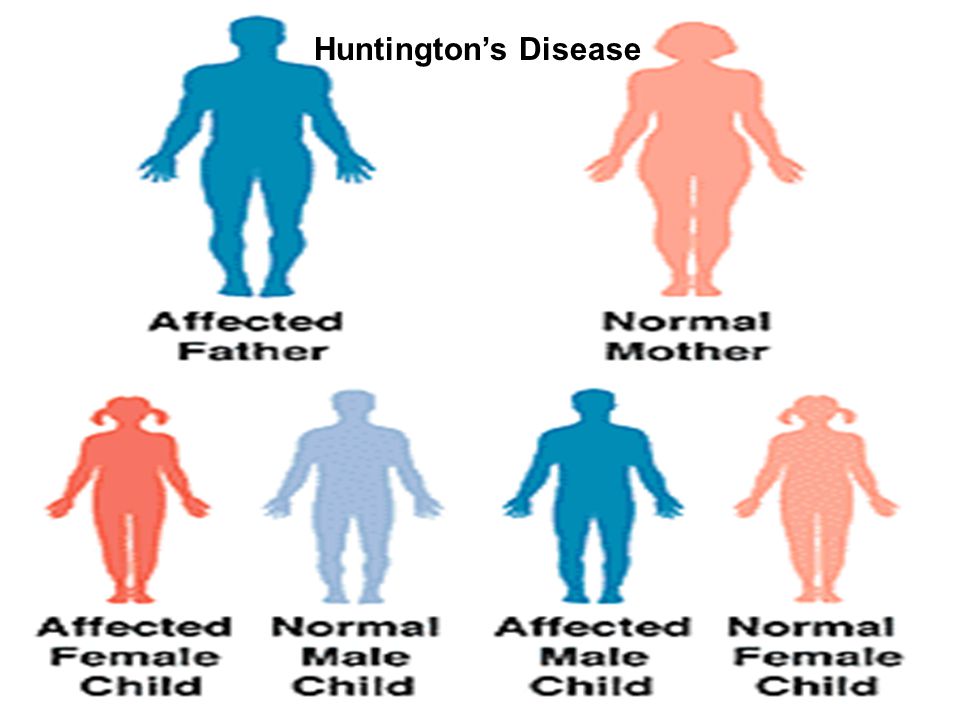 При этом у его родителя уже могут быть симптомы заболевания либо они разовьются позже в течение жизни.
При этом у его родителя уже могут быть симптомы заболевания либо они разовьются позже в течение жизни.
соответствующем гене. Таким образом, гены выступают в качестве схемы — последовательности инструкций для клетки о том, как построить определённые белки. Ген HTT содержит инструкции о том, как построить белок гентингтин. Белки — это молекулы, которые выполняют различные функции внутри клетки — они обеспечивают множество необходимых для неё процессов, таких как ферментативные реакции и поддержание структуры. Если белок функционирует неправильно или отсутствует, например, из-за увеличения в нём числа копий какого-то участка (экспансии), то это может повлиять на клетку и, в конечном счёте, на организм в целом, приводя в ряде случаев к развитию болезни.Что такое белок?

Белок гентингтин довольно крупный и образуется (или, по-научному, экспрессируется) с различной интенсивностью в каждой клетке организма человека, однако в наибольшей степени это происходит в головном мозге. По всей видимости, гентингтин является очень важным белком, т.к. его отсутствие приводит к смерти мышей ещё на эмбриональном этапе. На одном из концов молекулы гентингтина есть последовательность повторяющейся аминокислоты — глутамина. Этот особенный структурный участок, называемый полиглутаминовым повтором, как правило, содержит до 35 остатков глутамина. У людей, являющихся носителями мутации БГ, этот участок гентингтина содержит не менее 36 повторов, что приводит к нарушению функционирования белка.Белок гентингтин
БГ является доминантно наследуемым заболеванием.Каким образом наследуется БГ?
 Это означает, что человек, рождённый с одной мутантной копией гена HTT, заболеет БГ несмотря на наличие у него и одной нормальной копии этого гена. Носитель мутации БГ, вне зависимости от того, симптомный он или нет, может передать с 50 %-ной вероятностью как нормальную, так и мутантную копию гена (при условии, что у него/неё мутантной является только одна из двух копий гена HTT). Медицинские технологии позволяют сделать так, чтобы его/её детям передалась только здоровая копия гена HTT. С другой стороны, у человека, который не унаследовал мутантный ген HTT, БГ уже никогда не разовьётся, и его/её дети тоже будут вне риска относительно этого заболевания. Мутация БГ не может перепрыгнуть поколение. Тем не менее, может случиться, что носитель мутации умер до того, как у него/неё появились бы симптомы болезни, а его его/её дети не знают, что имеют риск развития БГ.
Это означает, что человек, рождённый с одной мутантной копией гена HTT, заболеет БГ несмотря на наличие у него и одной нормальной копии этого гена. Носитель мутации БГ, вне зависимости от того, симптомный он или нет, может передать с 50 %-ной вероятностью как нормальную, так и мутантную копию гена (при условии, что у него/неё мутантной является только одна из двух копий гена HTT). Медицинские технологии позволяют сделать так, чтобы его/её детям передалась только здоровая копия гена HTT. С другой стороны, у человека, который не унаследовал мутантный ген HTT, БГ уже никогда не разовьётся, и его/её дети тоже будут вне риска относительно этого заболевания. Мутация БГ не может перепрыгнуть поколение. Тем не менее, может случиться, что носитель мутации умер до того, как у него/неё появились бы симптомы болезни, а его его/её дети не знают, что имеют риск развития БГ.
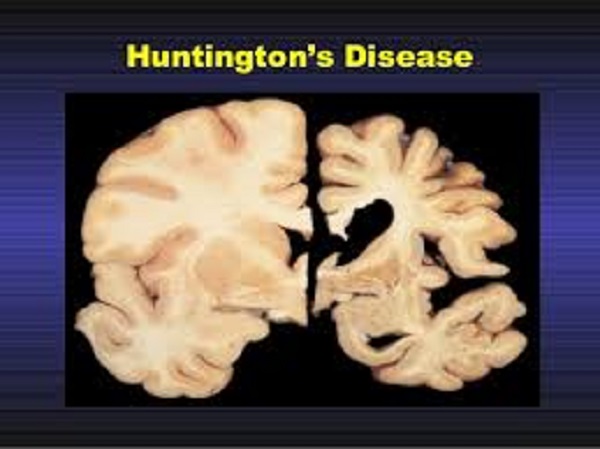
Определённые функции головного мозга, такие как контроль движений, мышление и речь, при БГ постепенно нарушаются, т. Полосатое тело представляет собой структуру, расположенную глубоко в центральной части головного мозга; при БГ оно поражается в первую очередь. Рисунок из Wikipedia.com.Участки головного мозга, поражающиеся при БГ
 к. ответственные за них нервные клетки повреждаются и гибнут. Часть головного мозга, которая страдает в наибольшей степени при БГ, носит название полосатое тело и является частью так называемых базальных ядер — структур, расположенных глубоко в центральной части головного мозга. В первую очередь, полосатое тело отвечает за планирование и контроль движений, но оно также обеспечивает и многие другие процессы, включая мышление и эмоции. По мере прогрессирования БГ повреждается и кора головного мозга (наиболее поверхностно расположенная его часть, имеющая извилины), что усугубляет выраженность когнитивных нарушений. В целом, с течением времени БГ приводит к атрофии всего головного мозга, приводя к общей инвалидизации человека.
к. ответственные за них нервные клетки повреждаются и гибнут. Часть головного мозга, которая страдает в наибольшей степени при БГ, носит название полосатое тело и является частью так называемых базальных ядер — структур, расположенных глубоко в центральной части головного мозга. В первую очередь, полосатое тело отвечает за планирование и контроль движений, но оно также обеспечивает и многие другие процессы, включая мышление и эмоции. По мере прогрессирования БГ повреждается и кора головного мозга (наиболее поверхностно расположенная его часть, имеющая извилины), что усугубляет выраженность когнитивных нарушений. В целом, с течением времени БГ приводит к атрофии всего головного мозга, приводя к общей инвалидизации человека.
информирование других членов семьи о том, что они тоже имеют риск носительства мутации этого заболевания.Как БГ влияет на повседневную жизнь?
 Взрослые люди молодого возраста в особенности должны иметь в виду последствия выявления мутации БГ для образования, обучения и трудоустройства. По мере прогрессирования болезни человек постепенно теряет возможность быть жить независимо от других. Трудовая деятельность, социальная жизнь и общие повседневные дела становятся проблематичными, и пациенты становятся всё более зависимы от помощи со стороны их родственников, работников здравоохранения и социальных работников. Более подробную информацию и помощь можно получить в местных пациентских организациях и по БГ.
Взрослые люди молодого возраста в особенности должны иметь в виду последствия выявления мутации БГ для образования, обучения и трудоустройства. По мере прогрессирования болезни человек постепенно теряет возможность быть жить независимо от других. Трудовая деятельность, социальная жизнь и общие повседневные дела становятся проблематичными, и пациенты становятся всё более зависимы от помощи со стороны их родственников, работников здравоохранения и социальных работников. Более подробную информацию и помощь можно получить в местных пациентских организациях и по БГ.
Эффективные способы приспособиться к жизни с БГ должны быть индивидуальными — они зависят от самого человека, стадии заболевания и семейного контекста. БГ развивается очень медленно, что, в целом, даёт время для приспособления к тем изменениям, что она приносит.Есть ли способы лучше приспособиться к жизни с БГ?
 В разработке стратегии выстраивания жизни с учётом этих изменений и в поддержании хороших отношений с человеком, страдающим БГ, ухаживающим лицами и близким людям может помочь более детальное понимание поведенческих и когнитивных нарушений, которые несёт заболевание. Полезную информацию и советы можно получить у и в пациентских организациях.
В разработке стратегии выстраивания жизни с учётом этих изменений и в поддержании хороших отношений с человеком, страдающим БГ, ухаживающим лицами и близким людям может помочь более детальное понимание поведенческих и когнитивных нарушений, которые несёт заболевание. Полезную информацию и советы можно получить у и в пациентских организациях.
На вы найдёте список языковых координаторов, которые могут вам помочь. Вы также можете воспользоваться формой для обратной связи, расположенной по этой ссылке.Как я могу связаться с EHDN?
Вы можете записаться на консультацию, либо получив направление от вашего врача общей практики, либо связавшись с от EHDN для вашей страны, который подскажет вам, к кому можно обратиться.Как я могу получить консультацию специалиста?

Независимой мнение по БГ можно получить в пациентской организации в вашей стране.Есть ли возможность поговорить со специалистом, не посещая
больницу?
EHDN играет ключевую роль в исследовании мирового масштаба Enroll-HD. Это наблюдательное исследование, которое не подразумевает изучения действия каких-либо вмешательств. Таким образом, само по себе оно не предполагает исследования экспериментальных методов лечения. Участники исследования проходят клиническое обследование в рамках ежегодных визитов; на основании получаемых данных участники могут подходить для участия в клинических исследованиях симптоматических или болезнь-модифицирующих методов лечения, если таковые появляются. Участие в Enroll-HD доступно на базе многих исследовательских центров по БГ в разных странах мира. Выяснить, есть ли такой центр вблизи вашего места проживания, можно здесь либо у по вашей стране, который сможет проинформировать вас об исследованиях, проводимых в вашем регионе. Ваша местная пациентская организация(и) сможет(гут) предоставить вам общую информацию об участии в исследованиях. Для более подробной информации об изучении БГ пройдите, пожалуйста, по или посетите сайт HDBuzz, на котором вы сможете найти новости об исследовании БГ, написанные учёными понятным языком и переведённые на большинство языков.Как я могу присоединиться к изучению БГ?

Да, ряд организаций по защите прав пациентов предоставляет поддержку людям и семьям, связанным с БГ.Существуют ли группы поддержки, специализирующиеся на БГ?
 Связаться с этими организациями можно через вашего врача общей практики или специалиста по БГ; вы можете также обратиться в эти организации сами напрямую. Европейская ассоциация по болезни Гентингтона (англ. European Huntington’s disease Association, (EHA) ведёт список пациентских организаций которые могут вам помочь.
Связаться с этими организациями можно через вашего врача общей практики или специалиста по БГ; вы можете также обратиться в эти организации сами напрямую. Европейская ассоциация по болезни Гентингтона (англ. European Huntington’s disease Association, (EHA) ведёт список пациентских организаций которые могут вам помочь.
По любым другим вопросам свяжитесь, пожалуйста, с вашим от EHDN или с местной пациентской организацией.
Что за болезнь хорея Гентингтона – причины развития, симптомы — клиника «Добробут»
Главная
Медицинская библиотека Добробут
Дата публикации: 2020-08-23
Хорея Гентингтона – этиология, симптомы. Лечение гиперкинезов у детей и взрослых
Болезнь хорея Гентингтнона (Хантингтона) относится к наследственным заболеваниям и характеризуется тяжелым поражением центральной нервной системы. Причиной ее развития являются патологические изменения в геноме человека. Поражение может коснуться не только коры головного мозга, но и его мягких тканей. Заболевание протекает тяжело, активно прогрессирует.
Поражение может коснуться не только коры головного мозга, но и его мягких тканей. Заболевание протекает тяжело, активно прогрессирует.
Симптомы патологии
Современная медицина выделила две основные формы болезни:
- классическая хорея Гентингтона – прогрессирование начинается после 40 лет, клиническая картина характеризуется снижением мышечного тонуса и избыточными движениями;
- ювенильная форма – проявляется в детском возрасте, составляет 10% всех случаев заболевания.
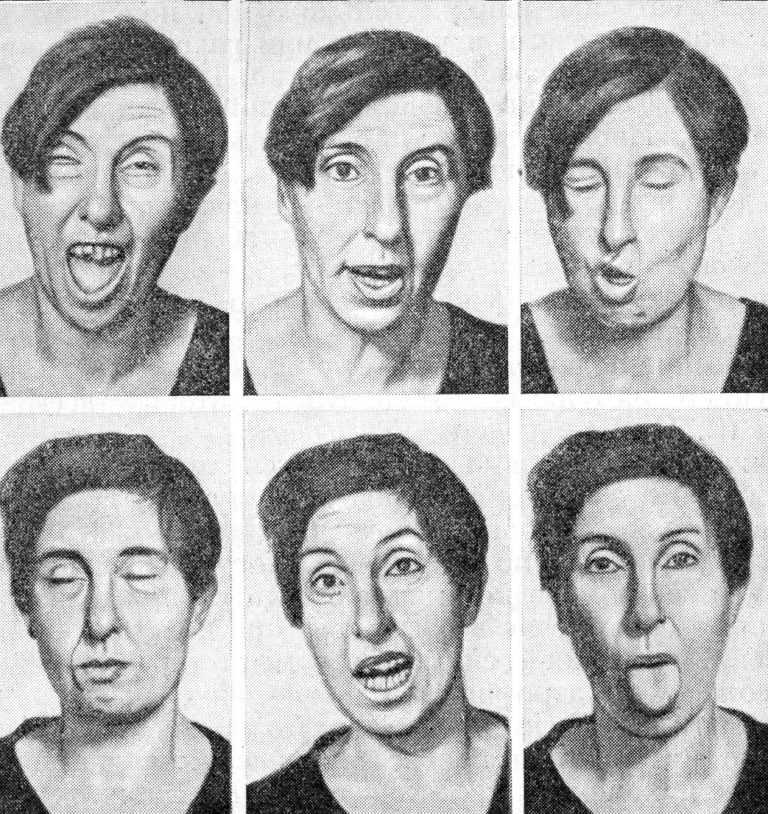
Развитие болезни быстрое, но поэтапное. Симптомы малой хореи у детей:
- причмокивание губами во время разговора;
- частые и глубокие вздохи;
- постоянное шмыганье носом;
- спонтанное высовывание языка.
Мимические самопроизвольные движения на лице постепенно становятся интенсивнее, к ним присоединяется приплясывание, кивание головой не к месту, размахивание руками, шатание корпусом из стороны в сторону. Ребенку будет поставлен диагноз тикозный гиперкинез и рекомендовано наблюдение невропатолога.
Ребенку будет поставлен диагноз тикозный гиперкинез и рекомендовано наблюдение невропатолога.
По мере роста ребенка вышеуказанные симптомы могут исчезать, но отмечается общая замедленность в поведении. Впоследствии мышечный тонус становится максимально высоким и больной не в состоянии даже сам себя обслужить – застегнуть пуговицы, надеть брюки и так далее.
Ярким симптомом хореи Гентингтона является нарушение психики – сначала это повышенная вспыльчивость и эмоциональность, нарушения сна. По мере прогрессирования патологии проявляется немотивированная агрессия, а в более старшем возрасте отмечаются галлюцинации, бредовые идеи и даже суицидальные мысли. При этом критично относиться к собственному состоянию и поведению больные не могут и считают себя абсолютно здоровыми.
Результатом нарушения генома становится слабоумие и деменция, когда человек нуждается в постоянном уходе. Прогрессирование заболевания длится на протяжении 10-20 лет. Ювенальная форма хореи Гентингтона развивается быстро, летальный исход происходит через 8-10 лет.
Лечение хореи Гентингтона
Современная медицина не имеет четкой тактики терапии, это заболевание считается неизлечимым. В отдельных в качестве лечения гиперкинезов у детей назначаются:
- Тетмодис – уменьшает двигательные нарушения;
- нейролептики (Галоперидол, Азалептин и другие) – снижают мышечный тонус, тормозят прогрессирование нарушений психики;
- Амитриптилин – антидепрессант, снижающий интенсивность двигательных и мимических нарушений.
Других терапевтических назначений не делается, потому что любое вмешательство не дает каких-либо результатов. Больные буквально обречены, потому что в результате они становятся совсем неуправляемыми, не ориентируются в пространстве и времени, теряют память и не узнают никого из родных. Они должны находиться под постоянным контролем, поэтому чаще всего помещаются в специализированные психиатрические клиники.
Хорея Гентингтона – это нарушение генетики. Поэтому при появлении такого больного в роду, необходимо пройти обследование всем членам семьи, а планирование беременности лучше проводить под строгим контролем генетиков. Современные ученые могут предложить людям, попадающим в группу риска по рождению такого малыша, пройти процедуру ЭКО или ИКСИ – можно будет подсадить матери здоровую оплодотворенную яйцеклетку.
Современные ученые могут предложить людям, попадающим в группу риска по рождению такого малыша, пройти процедуру ЭКО или ИКСИ – можно будет подсадить матери здоровую оплодотворенную яйцеклетку.
Более полную информацию о патологии, о заболевании ревматическая хорея можно узнать на страницах нашего сайта www.dobrobut.com.
Связанные услуги:
Консультация невролога
Хотите получить онлайн разъяснение от врача МС “Добробут”?
Скачивайте наше приложение Google Play и App Store
Наши врачи
Смотреть всех врачей 634
Наши сертификаты
Сертифікат № QIZ 804 468 C1
Сертифікат № QIZ 804 469 C1
Сертифікат № QIZ 804 470 C1
Сертифікат № QIZ 804 471 C1
Смотреть все сертификаты
Заказать обратный звонок
Введите Ваш телефон
Другие статьи
Что такое эндометриоз, первые признаки и диагностика. Как лечат эндометриоз
Как лечат эндометриоз
Внутренний и наружный эндометриоз. Симптомы, которые могут указывать на развитие патологии, особенности лечения медикаментами и народными средствами.
Виды наркоза – спинальная и эпидуральная анестезия, эндотрахеальный и местный наркоз
Какие виды наркоза бывают. В чем преимущества местного и общего наркоза. Особенности спинального и эпидурального наркоза. Возможные последствия наркоза
Гемангиома кожи у детей: причины возникновения, лечение, прогноз
Что такое гемангиома. Виды гемангиом. Локализация сосудистых опухолей. Причины возникновения гемангиомы губы. Основные методы лечения гемангиом. Операция по удалению гемангиомы лазером.
Первые симптомы гипертонического криза, причины и лечение неотложного состояния
Гипертонический криз – это тяжелое состояние, развивающееся при резком повышении АД. К осложнениям гипертонического криза относятся поражение мозга, почек и сердца. При гипертоническом кризе необходимо оказать неотложную помощь
К осложнениям гипертонического криза относятся поражение мозга, почек и сердца. При гипертоническом кризе необходимо оказать неотложную помощь
Смотреть все статьи
Заказать обратный звонок
Введите Ваш телефон
причины, симптомы и лечение в статье невролога Гольченко Е. А.
Над статьей доктора Гольченко Евгении Александровны работали литературный редактор Юлия Липовская, научный редактор Надежда Гавран и шеф-редактор Лада Родчанина
Дата публикации 27 ноября 2020Обновлено 26 апреля 2021
Определение болезни. Причины заболевания
Болезнь Гентингтона (хорея Гентингтона) — это хроническое, неуклонно прогрессирующее, наследственное аутосомно-доминантное заболевание нервной системы. Болезнь проявляется нарушением речи, памяти и присутствием навязчивых вычурных движений, мешающих выполнять повседневную деятельность. Со временем пациент теряет навыки самообслуживания и нуждается в опеке, то есть инвалидизируется.
Болезнь проявляется нарушением речи, памяти и присутствием навязчивых вычурных движений, мешающих выполнять повседневную деятельность. Со временем пациент теряет навыки самообслуживания и нуждается в опеке, то есть инвалидизируется.
Термин «хорея» означает «танец» на греческом языке. Впервые это определение использовал известный алхимик Парацельс при описании «пляски святого Витта». Эта «пляска», возможно, была связана с религиозным экстазом, то есть имела истерический характер [1].
Позже английский врач Томас Сиденгам в процессе изучения «пляски святого Витта» описал детскую форму хореи, теперь это заболевание носит его имя. В 1872 году американский врач Джордж Гентингтон отметил наследственный вариант хореи с началом во взрослом возрасте. Впоследствии это заболевание получило название «болезнь Гентингтона» (БГ). Также его называют хореей Гентингтона (или Хантингтона), наследственной или дегенеративной хореей.
Классическим описанием патологии считается эссе «О хорее», опубликованное Джорджем Гентингтоном в «Медицинском и хирургическом вестнике» [2]. В настоящее время хорея Гентингтона является самой частой причиной наследственной хореи. Одним из самых известных людей, страдавших этим заболеванием, был американский кантри певец и композитор Вуди Гатри, который заболел в 50-е годы.
В настоящее время хорея Гентингтона является самой частой причиной наследственной хореи. Одним из самых известных людей, страдавших этим заболеванием, был американский кантри певец и композитор Вуди Гатри, который заболел в 50-е годы.
Болезнь может начинаться в разном возрасте, но чаще всего симптомы появляются в 30-50 лет и неуклонно прогрессируют [5]. Описаны две основные клинические формы болезни Гентингтона:
- гиперкинетическая, наиболее частая, характеризуется более поздним началом, постепенным прогрессированием хореических гиперкинезов (насильственных избыточных двигательных актов, возникающих помимо воли больного), а также деменцией с замедлением когнитивных функций, снижением критики, депрессией, разнообразными психозами;
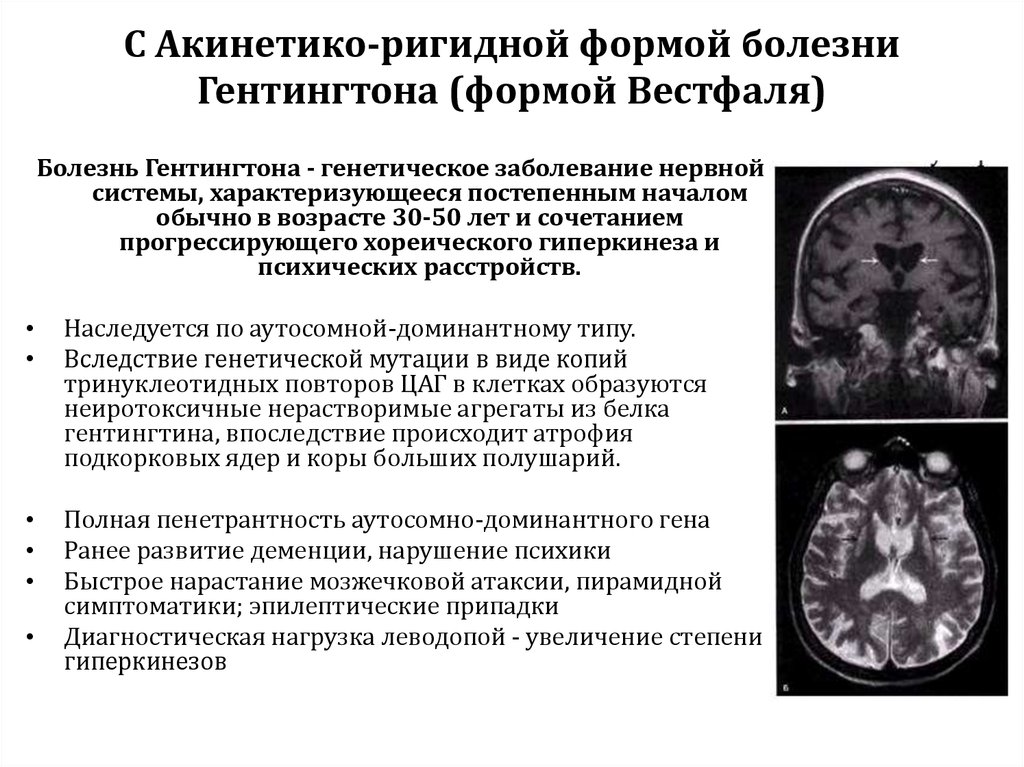
- акинетико-ригидная с началом в раннем возрасте (вариант Вестфаля) наблюдается в 5-10 % случаев, характеризуется быстрым нарастанием мышечной слабости, контрактур (ограничением подвижности суставов), нарушениями поведения и психики, эпилепсией, наблюдается при передаче мутантного гена от отца.

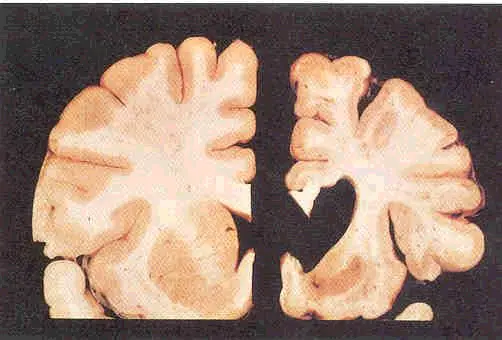
Нейроморфологическая картина характеризуется уменьшением, то есть атрофией, полосатого тела (стриатума — глубинной структурной части мозга) уже на самой ранней стадии заболевания. На более развёрнутой стадии возможна атрофия коры головного мозга.
Распространённость хореи в Западной Европе составляет в среднем 4-10 случаев на 100 000 населения, поэтому патологию можно отнести к редким заболеваниям [3]. Наименьшая частота хореи Гентингтона установлена среди жителей Японии, Финляндии, Южной Африки и Северной Америки (7,33 на 100 000 населения). Самая высокая частота (более 17 случаев на 100 000 населения) зафиксирована в районе озера Маракайбо в Венесуэле, в Тасмании и на островах Маврикий [4]. В Российской Федерации распространённость патологии в целом не оценивалась, цифры по различным регионам разнятся.
Мужчины и женщины болеют одинаково часто, однако существует механизм так называемой «отцовской передачи» заболевания, при котором наиболее тяжёлые варианты болезни наследуются от отца, особенно в случае передачи мутантного гена на протяжении нескольких поколений.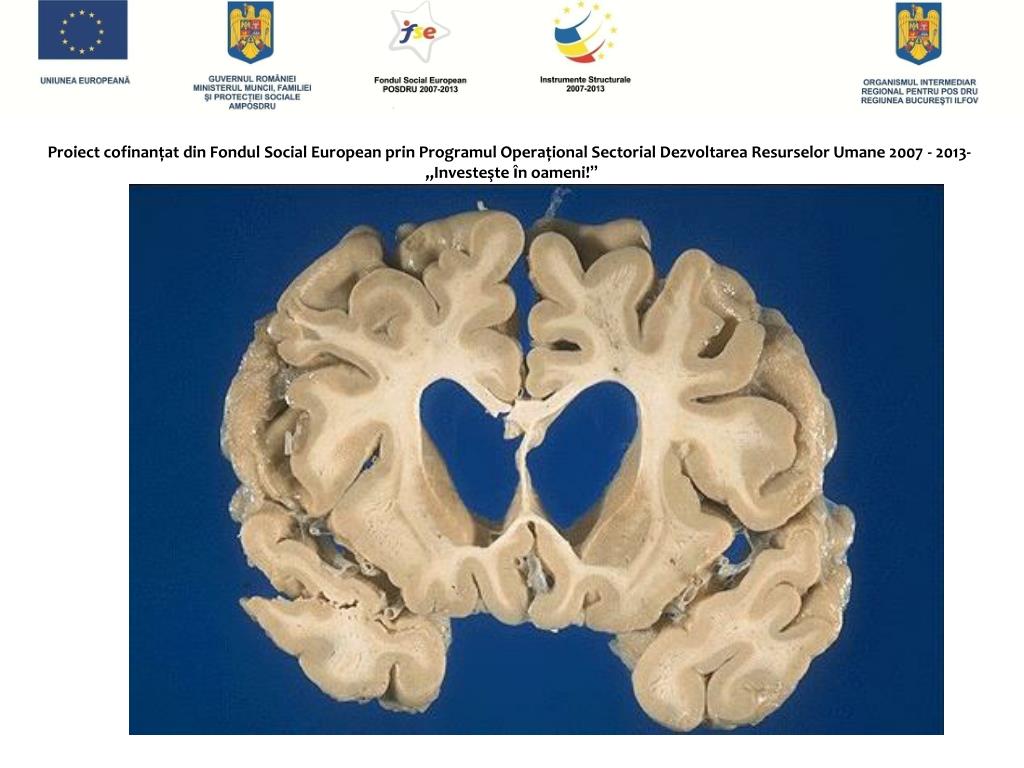 Это объясняется нестабильностью региона, содержащего мутацию, и нарастанием числа повторов в процессе сперматогенеза [31].
Это объясняется нестабильностью региона, содержащего мутацию, и нарастанием числа повторов в процессе сперматогенеза [31].
Причина заболевания — мутация в гене HTT, который отвечает за образование белка гентингтина. Гены — самые маленькие единицы наследственности, которые содержатся в хромосомах. В соматических клетках человека всего одна пара половых хромосом и 22 пары аутосом. Болезнь Гентингтона явлется аутосомно-доминантным заболеванием, то есть мутантный ген расположен в аутосоме и является преобладающим. Это значит, что для развития болезни достаточно унаследовать мутантный ген от одного из родителей.
Предрасполагающими факторами развития болезни могут стать психотравмы, черепно-мозговые травмы или перенесённые инфекции.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением — это опасно для вашего здоровья!
Симптомы хореи Гентингтона
При болезни Гентингтона присутствует многолетняя стадия «предболезни», которая определяется постепенным нарастанием субклинических и биохимических изменений в структурах головного мозга. В самом начале появляются двигательные нарушения: насильственные избыточные двигательные акты, возникающие помимо воли больного — хореический гиперкинез. С развитием болезни присоединяются когнитивные или психические расстройства.
В самом начале появляются двигательные нарушения: насильственные избыточные двигательные акты, возникающие помимо воли больного — хореический гиперкинез. С развитием болезни присоединяются когнитивные или психические расстройства.
Прогрессирование заболевания в некоторых случаях может сопровождаться постепенным уменьшением выраженности непроизвольных движений, но усилением психических симптомов [9]. Однако для каждой из форм болезни выделяют превалирующие симптомы.
Клинические проявления можно распределить по следующим группам [10]:
1. Двигательные нарушения могут проявляться общим двигательным беспокойством в движениях рук и ног, возникающим при стрессе, волнении или ходьбе. Патогномоничным (характерным) и самым ранним двигательным симптомом являются глазодвигательные нарушения. По мере прогрессирования заболевания появляются грубые нарушения следящих движений глазных яблок, следовательно, пациент практически не может зафиксировать взгляд на каком-либо предмете. Постепенно к хорее присоединяются иные нарушения, например дистония (стабильные рефлекторные двигательные сокращения отдельных групп мышц) и паркинсонизм (снижение скорости движения, скованность мышц и дрожание конечностей).
Постепенно к хорее присоединяются иные нарушения, например дистония (стабильные рефлекторные двигательные сокращения отдельных групп мышц) и паркинсонизм (снижение скорости движения, скованность мышц и дрожание конечностей).
Одним из основных двигательных нарушений является отсутствие контроля произвольных движений. Пациент не может удерживать какую-либо позу по причине грубых насильственных двигательных актов. На более поздних стадиях больным сложно выполнять движения, которые требуют тонкой координации. Значительно замедляется двигательная активность и нарушается ходьба, в результате человек часто падает. Больные широко расставляют ноги при ходьбе, корпус тела и шея вытянуты вперёд, корпус тела несколько повёрнут вокруг своей оси. При ходьбе появляется как бы пританцовывание, если пациент не пользуется компенсаторными приёмами, чтобы уменьшить насильственное движение (например, если не держится одной рукой за другую). Также повышается вероятность возникновения тонического напряжения мышц, приводящих бёдра. В таком случае больные передвигаются со сведёнными бёдрами и коленями.
В таком случае больные передвигаются со сведёнными бёдрами и коленями.
Виды гиперкинезов:
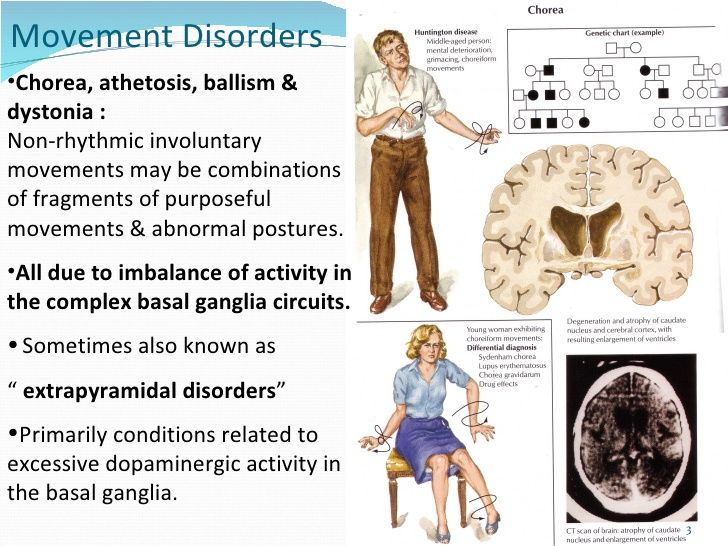
- Хорея — проявляется неритмичным, отрывистым, кажущимся произвольным движением.
- Атетоз — это гиперкинез, который проявляется замедленным и червеобразным движением.
- Баллизм — это резкие, размашистые движения рукой или ногой.
- Дистония характеризуется медленным и неритмичным мышечным сокращением, которое часто приводит к формированию патологической позы.
- Миоклонус — это внезапные, быстрые и отрывистые движения, напоминающее удар электрического тока.
Другие двигательные проявления:
- Брадикинезия (медлительность).
- Мышечная ригидность (скованность).
- Окуломоторные (глазодвигательные) расстройства.
- Падения.
2. Когнитивные нарушения. Трудности в общении, организации собственной деятельности, упорядочивании своих мыслей и в усвоении новой информации, а также нарушения восприятия, повышенная отвлекаемость, нарушения памяти и умственной работоспособности могут появляться уже на ранних этапах заболевания. Это практически универсальные проявления хореи Гентингтона. Максимально страдают исполнительные функции, то есть способность планировать свои действия, а также оценивать агрессивное поведение. У большинства пациентов отмечается замедление психомоторных процессов, появляется апатия, снижение внимания к себе.
Это практически универсальные проявления хореи Гентингтона. Максимально страдают исполнительные функции, то есть способность планировать свои действия, а также оценивать агрессивное поведение. У большинства пациентов отмечается замедление психомоторных процессов, появляется апатия, снижение внимания к себе.
3. Психические нарушения: депрессия, тревога, апатия, обсессивно-компульсивные расстройства (навязчивые и бредовые идеи), раздражительность, психотические расстройства (агрессия, галлюцинации и др.), гиперсексуальность. Психические нарушения при болезни Гентингтона характеризуются в первую очередь депрессией и тревогой. Нередко можно отметить раздражительность, которая иногда становится причиной агрессивного поведения. Появление обсессивно-компульсивных расстройств значимо ухудшает качество жизни родственников, проживающих вместе с больным. Значительно реже у пациентов могут появляться психозы. Реализованные суициды и суицидальные попытки встречаются при болезни Гентингтона в 4 раза чаще, чем в общей популяции, и в 5 % случаев становятся причиной гибели при этом заболевании [30].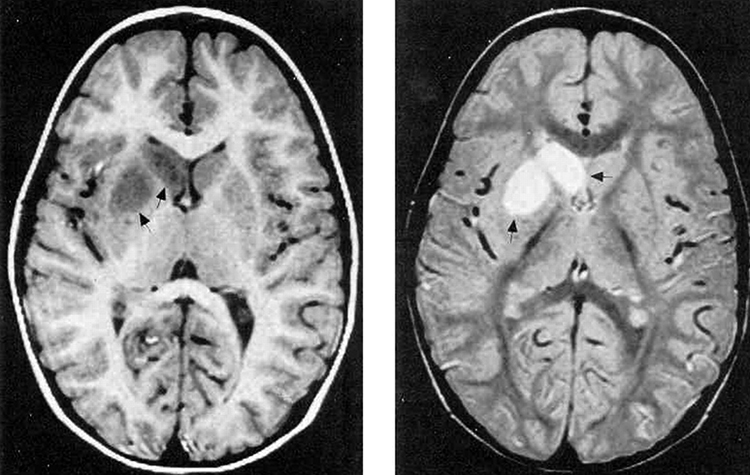
4. Метаболические расстройства: потеря массы тела, вплоть до кахексии (истощения) и др.
5. Прочие нарушения: расстройства сна, дизартрия (нарушение речи, при котором трудно произносить некоторые слова) и дисфагия (расстройство акта глотания). Больной испытывает трудности при разговоре, речь становится медленной, голос тихим и глухим, речь прерывается сопением, всхрапыванием, всхлипыванием, при осмотре выявляются элементы дизартрии. Тем не менее, даже на поздних стадиях пациенты доступны контакту. Из-за гиперкинезов языка, мягкого нёба, губ, диафрагмы и прямых мышц живота непроизвольные, «лишние» звуки (икота, сопение и др.) могут возникать даже когда больной молчит.
Патогенез хореи Гентингтона
Морфофункциональные изменения в мозге возникают намного раньше, чем появляются первые симптомы болезни Гентингтона [6][7]. Морфологические изменения в первую очередь касаются области полосатого тела. Характерным признаком при нейровизуализации является атрофия головки хвостатого ядра. В процесс вовлекаются: чёрная субстанция, кора больших полушарий, гиппокамп и мозжечок. Чуть в меньшей степени — боковые ядра гипоталамуса и таламус [8].
Характерным признаком при нейровизуализации является атрофия головки хвостатого ядра. В процесс вовлекаются: чёрная субстанция, кора больших полушарий, гиппокамп и мозжечок. Чуть в меньшей степени — боковые ядра гипоталамуса и таламус [8].
Патофизиологические механизмы развития болезни Гентингтона до конца не изучены. Большое значение в патогенезе этого заболевания имеет накопление и токсическое действие на нейроны мутантного белка гентингтина, в состав которого входит аминокислота глутамин.
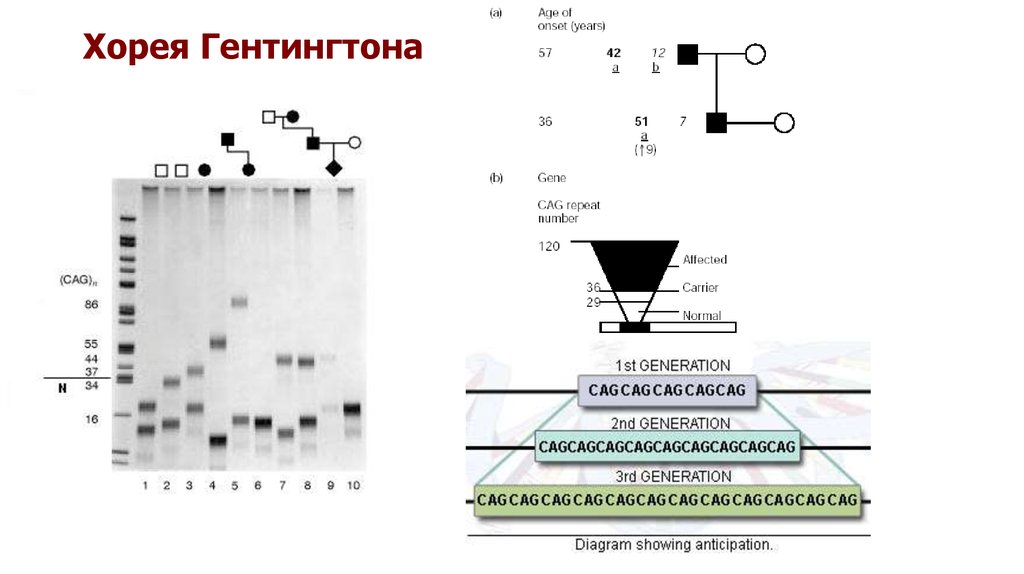
За образование белка гентингтина отвечает ген HTT, который расположен в коротком плече 4-й хромосомы человека. При отсутствии патологии в этом участке находится определённое количество повторов последовательности тринуклеотидов цитозин-аденин-гуанин (ЦАГ) — от 6 до 35. Мутация заключается в увеличении числа повторов ЦАГ (более 40), в результате чего мутантный белок гентингтин за счёт избыточного содержания остатков аминокислоты глутамина становится токсичным в отношении определённых видов нейронов [32].
Число повторов ЦАГ:
- До 26 — норма.
- От 27 до 35 (нестабильное количество) — симптомы отсутствуют, но есть повышенный риск того, что у детей будет болезнь Гентингтона.
- 36 или больше — патогенные мутации с различной пенетрантностью (проявление гена у 100 % лиц с соответствующим генотипом называется полной пенетрантностью, в остальных случаях — неполной пенетрантностью): 36-39 — «зона неполной пенетрантности»; более 40 — полная пенетрантность.
«Неправильный» белок повреждает нервные клетки головного мозга при встраивании в процесс обмена веществ. Поэтому возникают симптомы, характерные для болезни Гентингтона. Удлинённый участок молекулы белка гентингтина способствует формированию патологических межмолекулярных связей. Таким образом, болезнь Гентингтона является «полиглутаминовым заболеванием» [11]. Одним из новых патогенетических механизмов развития патологии, особенно в ранней стадии дегенеративного процесса, является нейровоспаление, активируемое мутантным гентингтином в центральной нервной системе через моноциты и микроглиальные клетки [12].
К особенностям наследования БГ относятся:
- Антиципация — феномен, при котором увеличение ЦАГ-повторов нарастает в ряду поколений, что означает более раннее возникновение болезни и более тяжёлое течение по сравнению с предыдущими поколениями.
- Феномен «отцовской передачи». Более тяжёлая форма заболевания развивается, если патология передалась от отца, симптомы появляются в детском возрасте.
- Наличие «зоны неполной пенетрантности», или «серой зоны» (у части лиц с такими мутациями болезнь развивается, а у других заболевание может отсутствовать даже в преклонном возрасте) [33].
- Эффект геномного импринтинга. При наследовании болезни Гентингтона гены отца и матери активированы или супрессированы по‐разному.
Интересно, что генетическая нестабильность мутации при болезни Гентингтона реализуется преимущественно у лиц мужского пола (процесс образования половых клеток), и эти наблюдения объясняют эффект «отцовской передачи» — манифестацию более ранних и тяжёлых случаев болезни гентингтона у потомков больного отца [13].
Классификация и стадии развития хореи Гентингтона
Для болезни Гентингтона, как и для других нейродегенеративных заболеваний, установлено существование длительной, многолетней стадии “предболезни”. В этот период действие мутантного гена ещё не успело привести к диагностируемой клинической картине, но патохимические нарушения в нейронах уже накапливаются, и возникает ряд субклинических проявлений [17].
Этапы жизни пациента с болезнью Гентингтона (классификация профессора неврологии Раймунда Рооса) [34]:
Доклиническая стадия (как правило, длится 10-15 лет до появления первых симптомов):
- Асимптомный период. У носителя мутации заболевания не отмечается субъективных, клинических или измеряемых инструментально изменений.
- Продромальный период (период предвестников заболевания) характеризуется постепенным появлением и нарастанием невыраженных двигательных, когнитивных и поведенческих изменений. Однако такие изменения не позволяют поставить клинический диагноз болезни Гентингтона
 Однако такие изменения не позволяют поставить клинический диагноз болезни Гентингтона
Однако такие изменения не позволяют поставить клинический диагноз болезни ГентингтонаКлиническая стадия (средняя продолжительность заболевания — 15-18 лет [35]):
- Клиническая стадия I. Появляются первые неврологические, когнитивные или психические симптомы. Основной симптом — беспорядочные, отрывистые, нерегулярные, кажущиеся произвольными движения. В повседневной жизни пациенту не требуется помощь. Летальный исход в этот период случается редко, за исключением случаев суицида.
- Клиническая стадия II. Нарастают двигательные нарушения, пациенту требуется помощь в самообслуживании. Смерть возможна в результате суицида.
- Клиническая стадия III. Тяжёлые двигательные нарушения, больной не способен на самообслуживание, полностью зависит от окружающих. Летальный исход наступает в результате осложнений заболевания.
На основании преобладания какого-либо синдрома различают несколько форм болезни Гентингтона:
1. «Классическая» гиперкинетическая форма. Характеризуется наличием навязчивых движений, нарушением походки (”танцующая походка”) и речи, а также снижением мыслительной деятельности.
«Классическая» гиперкинетическая форма. Характеризуется наличием навязчивых движений, нарушением походки (”танцующая походка”) и речи, а также снижением мыслительной деятельности.
2. Ригидная форма:
- Ювенильная (вариант Вестфаля). В отличие от классического варианта болезни, при ювенильной форме дебют происходит в возрасте до 21 года и наблюдается более тяжёлое течение и характер неврологической симптоматики. Проявления хореи минимальны, преобладают акинезия (отсутствие движения) и ригидность (болезненное состояние, при котором повышен тонус мышц и возникает сопротивление при попытке сделать движение). Кроме того, неврологические проявления при ювенильной форме более разнообразны: для неё характерны эпилепсия, атаксия (расстройство координации), миоклонии (молниеносные подёргивания отдельных групп мышц), дистония и другие расстройства, нетипичные для классической БГ. Давно известны генеалогические особенности этой формы: она развивается преимущественно при наследовании болезни от отца [16].
- Поздняя форма. Нередко является следствием прогрессирования «классической» формы болезни Гентингтона, при этом происходит трансформация клинической картины гиперкинетического синдрома в брадикинезию (замедленность движений) с мышечной скованностью. Примерно у 10 % взрослых пациентов болезнь начинается с акинетико-ригидного синдрома (движения скованные, трудно начать действие, медлительность во всем) с минимальными проявлениями хореи [15].
3. Психическая. Эта форма не является самостоятельной и выделяется лишь при резком преобладании психических симптомов над неврологической симптоматикой.
Чтобы оценить степень выраженности функциональных нарушений при патологии, разработана Унифицированная шкала оценки болезни Гентингтона (United Huntington’s Disease Rating Scale, UHDRS).
Компоненты UHDRS
- Двигательная оценка.
- Когнитивная оценка.
- Оценка поведенческих нарушений.
- Оценка функциональных возможностей.
- Оценка общей функциональной способности (TFC).

В зависимости от оценки по шкале общей функциональной способности (TFC), болезнь Гентингтона имеет следующую классификацию:
| Общий балл TFC | Стадия БГ | Общая характеристика |
|---|---|---|
| 11-13 | 1 | Ранние стадии: Появляются единичные двигательные нарушения: лёгкие неконтролируемые движения, незначительное изменение координации и походки, забывчивость, раздражительность. |
| 7-10 | 2 | |
| 4-6 | 3 | Стадия умеренных клинических проявлений: Присоединяются психические нарушения: нарушение речи и глотания, снижение аппетита и веса, становятся более выраженными неконтролируемые движения. |
| 1-3 | 4 | Поздние стадии, или стадии развёрнутых клинических проявлений: Появляются выраженные симптомы (двигательные и психические), пациент инвалидизируется, нуждается в посторонней помощи, появляются депрессия, галлюцинации, панические расстройства.  |
| 0 | 5 |
Осложнения хореи Гентингтона
- Аспирационная пневмония — воспаление которое развивается, когда в просвет лёгких попадают инородные вещества, например рвотные массы.
- Сепсис — опасное для жизни инфекционное заболевание человека, которое развивается в ответ на проникновение в кровь инфекционных агентов или их токсинов.
- Кахексия — истощение, которое возникает на фоне отказа от еды или нарушения глотания.
- Нарушения работы сердечно-сосудистой системы из-за закупорки артерий.
- Суицид в результате психических нарушений [10].
Диагностика хореи Гентингтона
Около 90 % диагнозов болезни Гентингтона, основанных на семейном анамнезе и выявлении характерных симптомов, подтверждаются генетическим исследованием. Поэтому сбор семейного анамнеза является обязательным при осмотре пациента с хореей.
Наиболее оптимальным методом диагностики является молекулярно-генетическое исследование. Для выявления мутации гена, ассоциированного с развитием хореи Гентингтона, требуется забор крови из вены. Анализ ДНК позволяет установить аномальное количество ЦАГ-повторов в гене HTT, что является непосредственной причиной развития болезни. С помощью этого метода можно подтвердить диагноз у больного и выявить носителей патологического гена среди родственников на доклинической стадии.
Аномальное количество ЦАГ-повторов в гене HTT не подтверждает диагноз, так как может быть получен задолго до появления первых симптомов. Нормальное количество ЦАГ-повторов в гене HTT говорит о том, что человек никогда не заболеет данной патологией.
При КТ головного мозга у пациентов с болезнью Гентингтона определяется расширение боковых желудочков и субарахноидальных пространств (между мозговыми оболочками, заполненными спинномозговой жидкостью). Характерно развитие гидроцефалии (избыточного скопления спинномозговой жидкости в желудочках головного мозга), при этом её степень будет зависеть от тяжести заболевания.
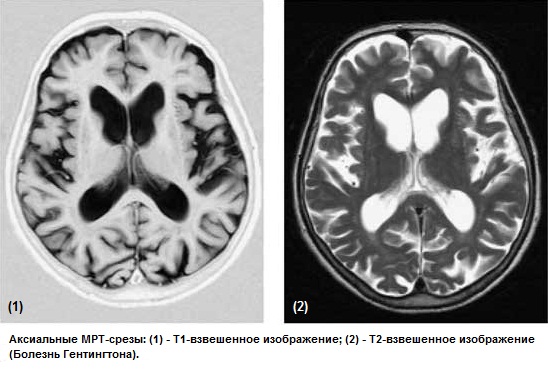
На МРТ можно отметить выраженную атрофию головок хвостатых ядер и расширение боковых желудочков, особенно в области передних рогов.
Однофотонная эмиссионная компьютерная томография (ОФЭКТ, англ. — SPECT) является более чувствительным и специфичным методом для постановки диагноза хореи Гентингтона: после введения радиофармпрепарата (РФП) выявляется снижение метаболизма глюкозы в хвостатых ядрах. Иногда эти изменения можно обнаружить на доклинической стадии болезни. Такие же признаки выявляются и при позитронно-эмиссионной томографии (ПЭТ) [18].
Количественная электроэнцефалография (ЭЭГ) — неинвазивный и относительно недорогой метод, в основе которого лежит регистрация электрической активности нейронов, которая представляет собой базовый механизм их взаимодействия. Ритмы ЭЭГ, в частности α- и θ-ритмы, отражают нейрофизиологические процессы, участвующие в обеспечении когнитивных функций [19][20].
Ритмы ЭЭГ:
- α-ритмы — регистрируется у человека в условиях физического и умственного покоя при закрытых глазах и отсутствии внешних раздражений.
- β-ритмы — регистрируются при открывании глаз, умственной работе и других раздражителях.
- θ-ритмы — наблюдается в состоянии неглубокого сна, при кислородном голодании мозга и наркозе.
- δ-ритмы — отмечаются во время глубокого сна.
Изменения на ЭЭГ у пациентов с болезнью Гентингтона характеризуются значимым снижением спектральной мощности α-ритма и повышением относительной спектральной мощности β- и δ-активности. Изучению данных ЭЭГ у асимптомных носителей мутаций в гене HTT посвящено небольшое количество работ, и их результаты неоднозначны.
Лабораторные анализы при болезни Гентингтона могут существенно не отличаться от нормы. При специальном биохимическом анализе крови иногда обнаруживается снижение концентрации некоторых компонентов:
- гамма-аминомасляной кислоты (ГАМК) — важнейшего тормозного нейромедиатора центральной нервной системы;
- глутаматдекарбоксилазы — фермента, необходимого для синтеза ГАМК;
- холин-ацетилтрансферазы — фермента, участвующего в синтезе нейромедиатора нервного возбуждения — ацетилхолина [21].

При исследовании спинномозговой жидкости нередко отмечается повышение содержания белка.
Дифференциальный диагноз. Существует ряд заболеваний, которые в некоторых случаях сопровождаются хореей с когнитивными и (или) психическими нарушениями и напоминают болезнь Гентингтона: болезнь Вильсона — Коновалова, болезнь Фридрейха, хореоакантоцитоз, спиноцеребеллярные атаксии и др.
Лечение хореи Гентингтона
Болезнь Гентингтона считается неизлечимым заболеванием, поэтому терапия является симптоматической и поддерживающей. Несмотря на это, благодаря лечению можно значительно улучшить качество жизни пациентов [22].
В настоящее время ведущую роль в лечении двигательных нарушений при болезни Гентингтона играет тетрабеназин. Это новый препарат, схожий по своим свойствам с атипичными нейролептиками [23]. Если нет выраженных психических проявлений болезни (в первую очередь депрессий, суицидальных мыслей, раздражительности, агрессивного поведения) и нарушения глотания, то этот препарат нужно рассматривать как средство первого выбора для лечения хореи при болезни Гентингтона.
В других случаях рационально рассмотреть возможность применения нейролептиков, т. е. антипсихотиков (в первую очередь атипичных — клозапин, кветиапин, рисперидон) в виде монотерапии или в сочетании с противоэпилептическими препаратами или бензодиазепинами [24].
Препараты вальпроевой кислоты (Депакин, Конвулекс и др.), известные как антиконвульсанты, применяются при наличии мышечных спазмов.
Коррекцию психических нарушений следует проводить при помощи лекарственных препаратов, применяемых в психиатрии.
По данным некоторых клинических случаев, могут быть эффективны высокие дозы клоназепама [25]. Этот короткодействующий препарат из группы бензодиазепинов чаще применяется в качестве вспомогательной терапии, если у пациента присутствует тревога. Кроме того, клоназепам назначается для коррекции миоклоний, дистонии и при нарушениях сна [26][27].
При необходимости врач назначает антидепрессанты, препаратами выбора являются ингибиторы обратного захвата серотонина благодаря небольшому числу побочных эффектов и отсутствию опасности передозировки (сертралин, эсциталопрам).
Пациентам с болезнью Гентингтона пытались проводить операции глубокой стимуляции мозга с имплантацией электродов во внутренний сегмент бледного шара (парной структуры переднего мозга, которая является перекрёстком выбора действия). При этом наблюдалось снижение выраженности хореических гиперкинезов, но отмечалось нарастание расстройств ходьбы, брадикинезии и когнитивных нарушений [28].
Есть данные о трансплантации в мозг пациентов с болезнью Гентингтона эмбриональных и стволовых клеток [29], но результаты такой экспериментальной терапии нуждаются в тщательной оценке.
В настоящее время в мире ведутся многоцентровые клинические исследования генной терапии у пациентов с хореей Гентингтона. Заболевание создаёт большие медико‐социальные проблемы, так как к моменту первых проявлений болезни люди успевают завести детей и передать им патологию, не подозревая об этом. В связи с этим необходима разработка новых методов диагностики и лечения, что делает актуальным дальнейшее изучения БГ и особенностей её течения.
Прогноз. Профилактика
Учитывая тот факт, что заболевание неуклонно прогрессирует, можно сделать вывод о неблагоприятном прогнозе. Средняя продолжительность жизни с момента появления гиперкинеза составляет 15-20 лет [15]. При ювенильной форме пациенты живут меньше.
Пациенты погибают не от самой болезни, а от её осложнений. Например, пневмония может стать смертельной из-за ослабленного иммунитета пациента. Иммунитет, в свою очередь, снижен из-за истощения пациента, который по причине проблем с глотанием мало принимает пищу. Травмы могут стать смертельными в результате нарушения движений и изменения походки. Частой причиной гибели пациентов является суицид [30].
Чем больше осведомлен пациент о своем заболевании, тем больше шансов своевременно получить необходимую терапию у врача невролога и предотвратить быстрое развитие болезни и осложнений.
Для профилактики заболевания в семьях с отягощённым анамнезом обязательно проведение медико-генетического консультирования для определения генетического статуса близких родственников и прогноза по заболеванию у потомства. Ввиду высокого риска повтора заболевания в семье с отягощённым анамнезом, в качестве профилактики рекомендуется проведение пренатальной диагностики или доимплантационного генетического тестирования эмбрионов (ПГТ-М), что обсуждается в рамках медико-генетического консультирования семьи.
Ввиду высокого риска повтора заболевания в семье с отягощённым анамнезом, в качестве профилактики рекомендуется проведение пренатальной диагностики или доимплантационного генетического тестирования эмбрионов (ПГТ-М), что обсуждается в рамках медико-генетического консультирования семьи.
Как спасти Тринадцатую? (Перспективы лечения болезни Хантингтона)
Статья на конкурс «био/мол/текст»: Сейчас сложно найти человека, который никогда не слышал про болезни Альцгеймера, Паркинсона или Хантингтона. Эти недуги относятся к группе нейродегенеративных заболеваний, вызывающих гибель нейронов и постепенное разрушение головного мозга. К сожалению, все они являются неизлечимыми. Поэтому ученые активно работают над тем, чтобы раскрыть механизмы развития этих болезней и найти терапию, которая поможет спасти пациентов. В своем исследовании мы обратились к пока еще малоизученному вопросу — что происходит с синаптической связью нейронов при нейродегенеративном процессе? Результаты этой работы открывают новое направление для разработки лекарства от болезни Хантингтона и других нейродегенеративных заболеваний.
Эта работа заняла первое место в номинации «Своя работа» конкурса «био/мол/текст»-2013.
Спонсор конкурса — дальновидная компания Thermo Fisher Scientific. Спонсор приза зрительских симпатий — фирма Helicon.
С увеличением средней продолжительности жизни все больше людей страдают от болезни Альцгеймера и болезни Паркинсона. К сожалению, годы исследований пока не привели ученых к открытию причин развития этих заболеваний и возможной терапии. Это связано, главным образом, с тем, что почти ничего не известно о факторах, вызывающих болезнь, а также с тем, что очень мало пациентов имеют генетическую предрасположенность. Чаще всего эти заболевания являются спорадическими, т.е. причины их возникновения не установлены. Это приводит к бесконечным спорам — никто не знает, как искусственно вызвать это заболевание у модельных животных для экспериментов и поиска лекарств. Поэтому все больше ученых обращают свое внимание на генетические заболевания нервной системы, такие как болезнь Хантингтона (БХ). Это заболевание также, как болезнь Альцгеймера и болезнь Паркинсона, относится к группе нейродегенеративных заболеваний, с которыми его объединяет ряд схожих черт: гибель нейронов центральной нервной системы, накопление амилоидоподобных агрегатов белков, когнитивные и двигательные нарушения у больных. При этом БХ имеет важное преимущество с точки зрения исследователей, т.к. известно, какая мутация вызывает это заболевание. Это дает возможность создавать точные генетические модели и исследовать их на животных. Это важно, потому что если мы поймем патогенез болезни Хантингтона, то нам легче будет разобраться и со спорадическими нейродегенеративными заболеваниями. Это мы и попытались сделать в своем исследовании.
Это заболевание также, как болезнь Альцгеймера и болезнь Паркинсона, относится к группе нейродегенеративных заболеваний, с которыми его объединяет ряд схожих черт: гибель нейронов центральной нервной системы, накопление амилоидоподобных агрегатов белков, когнитивные и двигательные нарушения у больных. При этом БХ имеет важное преимущество с точки зрения исследователей, т.к. известно, какая мутация вызывает это заболевание. Это дает возможность создавать точные генетические модели и исследовать их на животных. Это важно, потому что если мы поймем патогенез болезни Хантингтона, то нам легче будет разобраться и со спорадическими нейродегенеративными заболеваниями. Это мы и попытались сделать в своем исследовании.
Болезнь Хантингтона
Болезнь Хантингтона (БХ, в русскоязычной литературе также «болезнь Гентингтона») — наследственное заболевание нервной системы, которое поражает примерно 1 из 10 тыс. людей. Болезнь была впервые описана Джорджем Хантингтоном (George Huntington) в 1872, и с тех пор носит его имя, однако клинические симптомы этого заболевания были известны еще в XVI веке под названием «хорея» (от лат. choreus — танец). К признакам хореи относили непроизвольные, нескоординированные быстрые движения, похожие на судороги; именно так описывают и современные медики моторные нарушения, характерные для БХ. Болезнь может порой длиться до двадцати лет, но исход неизменно один и тот же: больной теряет способность самостоятельно передвигаться, говорить, а затем и мыслить. Как правило, симптомы болезни Хантингтона проявляются в возрасте от 30 до 50 лет, хотя у 5–10% пациентов отмечается появление симптомов в возрасте до 20 лет — так называемая ювенильная форма заболевания [1].
choreus — танец). К признакам хореи относили непроизвольные, нескоординированные быстрые движения, похожие на судороги; именно так описывают и современные медики моторные нарушения, характерные для БХ. Болезнь может порой длиться до двадцати лет, но исход неизменно один и тот же: больной теряет способность самостоятельно передвигаться, говорить, а затем и мыслить. Как правило, симптомы болезни Хантингтона проявляются в возрасте от 30 до 50 лет, хотя у 5–10% пациентов отмечается появление симптомов в возрасте до 20 лет — так называемая ювенильная форма заболевания [1].
Первый симптом болезни Хантингтона — непроизвольные подёргивания конечностей, торса и лицевых мышц. Довольно часто они сопровождаются резкими сменами настроения, депрессией, раздражительностью, неразборчивостью речи и неуклюжестью движений. По мере прогрессирования болезни, к этим симптомам добавляются затруднения или боль при глотании, неустойчивость походки, потеря равновесия, нарушение мыслительных функций и ухудшение памяти. В конце концов, больной теряет способность передвигаться без помощи посторонних и умирает обычно от пневмонии, остановки сердца или других осложнений.
В конце концов, больной теряет способность передвигаться без помощи посторонних и умирает обычно от пневмонии, остановки сердца или других осложнений.
Важной для врачей и исследователей особенностью БХ является то, что это заболевание является наследственным и вызывается мутацией в одном-единственном гене. Оказалось, что к развитию БХ приводит увеличение количества повторов триплета CAG, кодирующего глутамин, в первом экзоне гена белка хантингтина. При этом, чем больше количество повторов этого триплета, тем раньше начинается развитие заболевания. В норме в человеческой популяции встречается от 10 до 35 повторов. У пациентов с БХ количество повторов может быть от 36 до 121, при ювенильной форме — от 50 и выше [2]. Благодаря выявлению генетической основы заболевания, диагностика БХ в настоящее время не представляет проблемы; кроме того, возможной стала пренатальная диагностика заболевания и проверка эмбрионов перед имплантацией при ЭКО, которая позволяет иметь здоровых детей даже носителям мутантного гена.
К сожалению, выявление точной мутации все еще не позволяет ученым определить причину развития болезни Хантингтона и найти соответствующее лечение. Появление в клетке мутантного гена и, соответственно, измененного (мутантного) белка может привести к развитию патологии двумя путями: потеря функции (loss-of-function) или приобретение функции (gain-of-functin). В первом случае мутантный белок не может выполнять ту же функцию, что белок нормальный, и это приводит к нарушению клеточных процессов. Во втором случае, мутантный белок мешает нормальной жизнедеятельности клетки, начиная выполнять какую-то «лишнюю функцию». Чтобы разобраться, что происходит при БХ, ученые интенсивно изучают как функцию нормального белка хантингтина, так и поведение его мутантной формы [3].
К сожалению, попытки определить точную клеточную функцию хантингтина пока не увенчались успехом. Различные исследования указывают на участие этого белка в широком спектре биологических процессов, включая транспорт белков и везикул (мембранных пузырьков-транспортеров), организацию цитоскелета, клатрин-опосредованный эндоцитоз, постсинаптический сигналинг, регуляцию транскрипции и анти-апоптотические процессы [4]. Если удастся доказать, что нарушение какой-либо из этих функций является ключевым для развития заболевания, то лекарственные препараты для поддержания этой функции могут спасти пациентов с болезнью Хантингтона.
Если удастся доказать, что нарушение какой-либо из этих функций является ключевым для развития заболевания, то лекарственные препараты для поддержания этой функции могут спасти пациентов с болезнью Хантингтона.
Если верна гипотеза о приобретении функции, особое внимание стоит обратить на поведение мутантной формы хантингтина. Оказалось, что мутантный белок формирует агрегаты, которые являются одной из характерных черт развития БХ как у людей,так и у модельных животных (см.врезку). Сначала агрегаты были описаны только в ядре, однако последующие работы выявили их также в цитоплазме и отростках нейронов [5]. В последние годы многие авторы склоняются к тому, что образование агрегатов несет скорее протективную функцию, а основной патогенной формой мутантного хантингтина является мономерный растворимый белок [6].
Модели для изучения болезни Хантингтона
Модели БХ на животных появились более 30 лет назад. Первыми были модели, основанные на введении в стриатум нейротоксических веществ (например, хинолиновой кислоты — агониста NMDA-рецепторов), которые вызывали гибель нейронов. В настоящее время большинство исследователей работает на моделях трансгенных животных, среди которых есть не только мыши и крысы, но и беспозвоночные животные — мушка Drosophila melanogaster и червь Caenorhabditis elegans.
В настоящее время большинство исследователей работает на моделях трансгенных животных, среди которых есть не только мыши и крысы, но и беспозвоночные животные — мушка Drosophila melanogaster и червь Caenorhabditis elegans.
Мышиные модели болезни Хантингтона отличаются друг от друга количеством CAG-повторов и уровнем экспрессии трансгена — искусственно внесенного гена хантингтина. Т.к. именно от этих факторов зависит развитие БХ, разные линии мышей отличаются друг от друга скоростью развития патологий. К наиболее широко используемым моделям относят линии мышей R6/2, R6/1 и YAC128, которые были использованы и в нашей работе. У мышей этих линий симптомы заболевания наиболее выражены и проявляются достаточно быстро. Кроме того, у этих животных с возрастом прогрессируют когнитивные и моторные нарушения, развивается частичная потеря нейронов в стриатуме и коре.
Еще одним из способов моделирования БХ является использование клеточной культуры. В самом простом случае используются культуры клеток со стабильной трансфекцией гена хантингтина. Например, это клетки линии PC12, содержащие индуцибельный трансген первого экзона хантингтина или нейроны стриатума с экспрессией фрагментов хантингтина разной длины. Кроме того, можно использовать первичные культуры из нейронов трансгенных мышей или иммортализованные нейроны.
Например, это клетки линии PC12, содержащие индуцибельный трансген первого экзона хантингтина или нейроны стриатума с экспрессией фрагментов хантингтина разной длины. Кроме того, можно использовать первичные культуры из нейронов трансгенных мышей или иммортализованные нейроны.
Почему мы решили исследовать параметры синаптической передачи при болезни Хантингтона
Синаптическая передача — это передача сигналов между нейронами с помощью синаптического контакта. При возбуждении одного нейрона его синаптическое окончание выделяет в синаптическую щель медиатор — химическое вещество, которое оказывает свое возбуждающее или тормозящее воздействие на синаптическое окончание второго нейрона (рис. 1). Таким образом, синапсы связывают нейроны между собой, обеспечивая нормальное функционирование нейронных сетей и всей нервной системы. Если какая-то из систем головного мозга перестает функционировать, причина может крыться либо в нарушении работы отдельных нейронов, либо в нарушении связи между ними, т. е. нарушении синаптической передачи.
е. нарушении синаптической передачи.
Рисунок 1. Схематическое изображение устройства синапса.
«Википедия»
При болезни Хантингона поражается специфическая область головного мозга, называемая стриатумом. Стриатум является частью важного нейронного пути — экстрапирамидной системы, которая участвует в управлении движением и поддержании мышечного тонуса. Гибель нейронов стриатума при болезни Хантингтона приводит к разрушению экстрапирамидной системы, что связано с потерей контроля над движениями у больного человека. Но когда возникают первые патологические симптомы (тремор, нарушение координации), головной мозг человека еще не поврежден: нейроны начинают погибать только через несколько лет после начала развития заболевания. Т.е. болезнь начинается, когда что-то меняется в работе самих нейронов или в синаптической передаче, и эти нарушения впоследствии ведут к гибели нейронов и необратимым последствиям.
Накопившиеся за последние годы результаты исследований заставляют многих ученых склоняться к мысли, что именно нарушение нормальной работы системы нейрональной связи, синапсов и синаптической передачи, приводит к ранним нарушениям в работе экстрапирамидной системы. Оказалось, что в нейронах с мутациями в гене, кодирующем белок хантингтин, наблюдается целый ряд патологических изменений, которые нарушают синаптическую передачу. В таких мутантных клетках нарушается формирование и обновление запаса везикул (пузырьков с медиатором), изменяется внутриклеточная концентрация кальция , который необходим для нормального высвобождения медиатора в синаптическую щель, снижается количество ряда необходимых для функционирования синапса белков и т.п. [7]. Все это приводит к сниженному выбросу медиатора в синаптическую щель, а если медиатора недостаточно, то нейроны начинают хуже «слышать» друг друга, и команды, посылаемые корой головного мозга, не будут выполняться по всей строгости.
Оказалось, что в нейронах с мутациями в гене, кодирующем белок хантингтин, наблюдается целый ряд патологических изменений, которые нарушают синаптическую передачу. В таких мутантных клетках нарушается формирование и обновление запаса везикул (пузырьков с медиатором), изменяется внутриклеточная концентрация кальция , который необходим для нормального высвобождения медиатора в синаптическую щель, снижается количество ряда необходимых для функционирования синапса белков и т.п. [7]. Все это приводит к сниженному выбросу медиатора в синаптическую щель, а если медиатора недостаточно, то нейроны начинают хуже «слышать» друг друга, и команды, посылаемые корой головного мозга, не будут выполняться по всей строгости.
В 2013 году Нобелевской премии по физиологии и медицине удостоены работы, благодаря которым стали ясны детали везикулярного транспорта — процесса образования и транспортировки мембранных пузырьков (везикул) между клетками: «Нобелевская премия по физиологии и медицине (2013): везикулярный транспорт» [8]. — Ред.
— Ред.
Изучение нарушенной синаптической передачи при БХ было темой нашего исследования. Может ли быть, что неправильная работа нейронов стриатума на ранней стадии БХ вызвана тем, что они «не слышат» команды нейронов коры? Может ли ослабление синаптической связи приводить к необратимым изменениям в нейронах стриатума и вести к их гибели? О чем мы узнали во время поиска ответов на эти вопросы, рассказано ниже.
Результаты исследований: изменения в синаптической передаче при болезни Хантингтона
Изучать синаптическую передачу можно различными способами. Например, это можно делать, проникнув в нейронную цепочку с помощью методов электрофизиологии. Нейрон проявляет свою активность с помощью электрического тока, который можно измерить. Если экспериментатор возьмет цепочку из двух нейронов и, активировав один нейрон, зарегистрирует электрическую активность второго, он сможет выяснить, насколько хорошо проходит сигнал. Другой способ изучать функционирование синаптической передачи — исследовать морфологию нейрона. Дело в том, что многие нейроны (в том числе, нейроны коры и стриатума) имеют особые выросты мембраны — шипики, которые нужны им именно для образования синапсов (рис. 2). Чем более активно нейрон «общается» со своими соседями, тем больше на его поверхности шипиков. Взяв на вооружение эти два подхода, мы решили исследовать, как работает синаптическая передача при БХ.
Дело в том, что многие нейроны (в том числе, нейроны коры и стриатума) имеют особые выросты мембраны — шипики, которые нужны им именно для образования синапсов (рис. 2). Чем более активно нейрон «общается» со своими соседями, тем больше на его поверхности шипиков. Взяв на вооружение эти два подхода, мы решили исследовать, как работает синаптическая передача при БХ.
Рисунок 2. Дендритные шипики на поверхности стриатного нейрона. Шипики — небольшие выросты на поверхности нейрональных отростков; на увеличенном изображении они отмечены стрелками.
фото автора статьи
Рисунок 3. Нейрональная культура из нейронов коры и стриатума. С помощью специфических антител нейроны коры окрашены в красный цвет, а нейроны стриатума — в желто-зеленый.
фото автора статьи
В качестве модели для изучения болезни Хантингтона была использована клеточная культура из нейронов коры и стриатума. Для приготовления культуры незрелые нейроны из изучаемых зон мозга мышей высаживаются в чашки Петри, где они формируют полноценные нейрональные отростки и нейронные цепочки (рис. 3). Использовались мыши дикого типа (без мутаций) и мыши линии YAC128, которые несут мутацию в гене белка хантингтина и являются признанной моделью БХ. На 14–15 день после высаживания нейронов в чашку Петри они достигают зрелого состояния, соответствующего состоянию нейронов в мозге взрослого человека, а на 19–20 день нейроны считаются «старыми»: в них наблюдается ряд клеточных процессов, характерных для мозга пожилых людей. Кроме того, с возрастом в нейронах мышей YAC128 происходит накопление мутантного белка хантингтина и его агрегатов, поэтому изучение нейрональной культуры на этих двух этапах дает представление о том, что происходи в мозге пациента с БХ на ранней и на поздней стадиях заболевания.
3). Использовались мыши дикого типа (без мутаций) и мыши линии YAC128, которые несут мутацию в гене белка хантингтина и являются признанной моделью БХ. На 14–15 день после высаживания нейронов в чашку Петри они достигают зрелого состояния, соответствующего состоянию нейронов в мозге взрослого человека, а на 19–20 день нейроны считаются «старыми»: в них наблюдается ряд клеточных процессов, характерных для мозга пожилых людей. Кроме того, с возрастом в нейронах мышей YAC128 происходит накопление мутантного белка хантингтина и его агрегатов, поэтому изучение нейрональной культуры на этих двух этапах дает представление о том, что происходи в мозге пациента с БХ на ранней и на поздней стадиях заболевания.
Рисунок 4. Шипики разных типов на поверхности дендрита — микрофотография и схематическое изображение.
Для начала мы исследовали морфологические отличия между двумя линиями, т.е. сравнили их внешний вид. Для нормальных нейронов стриатума характерно наличие большого количества шипиков, благодаря чему их называют средними шипиковыми нейронами (СШН). Именно шипики формируют большую часть синаптических контактов между нейронами стриатума и коры, и для осуществления нормальной синаптической передачи важно наличие определенного их количества. Так же важно и «качество» шипиков: в современной нейробиологии их разделяют на три группы согласно размеру и форме (рис. 4): грибовидные, тонкие и пеньковые. При этом шипики разных типов выполняют разные функции: считается, что только грибовидные шипики формируют активные синапсы, в то время как тонкие и пеньковые контактов с другими нейронами не образуют. Таким образом, для нормального функционирования нейронной цепочки и эффективной передачи информации по ней необходимо наличие определенного количества грибовидных шипиков.
Именно шипики формируют большую часть синаптических контактов между нейронами стриатума и коры, и для осуществления нормальной синаптической передачи важно наличие определенного их количества. Так же важно и «качество» шипиков: в современной нейробиологии их разделяют на три группы согласно размеру и форме (рис. 4): грибовидные, тонкие и пеньковые. При этом шипики разных типов выполняют разные функции: считается, что только грибовидные шипики формируют активные синапсы, в то время как тонкие и пеньковые контактов с другими нейронами не образуют. Таким образом, для нормального функционирования нейронной цепочки и эффективной передачи информации по ней необходимо наличие определенного количества грибовидных шипиков.
Для того, чтобы узнать, сколько грибовидных шипиков должно быть на СШН в норме, на всех этапах морфологического анализа как контроль использовалась культура нейронов из головного мозга мышей дикого типа. Оказалось, что количество шипиков на СШН стриатума на 14 день культивирования (молодые нейроны) одинаково в культурах YAC128 и дикого типа, но на 20 день (у «старых» нейронов) наблюдаются значительные изменения (рис. 5). В «постаревшей» культуре YAC128 снижается общее количество шипиков, причем относительное количество грибовидных шипиков, которые образуют активные синапсы, уменьшается в два раза [10]. Получается, что морфологические изменения нейронов, свидетельствующие о нарушении синаптической передачи, развиваются только к «старости» (на поздней стадии заболевания). Значит, на ранних этапах корень проблемы должен лежать в другой области.
5). В «постаревшей» культуре YAC128 снижается общее количество шипиков, причем относительное количество грибовидных шипиков, которые образуют активные синапсы, уменьшается в два раза [10]. Получается, что морфологические изменения нейронов, свидетельствующие о нарушении синаптической передачи, развиваются только к «старости» (на поздней стадии заболевания). Значит, на ранних этапах корень проблемы должен лежать в другой области.
Рисунок 5. Морфологический анализ нейронов стриатума. а — шипики нейронов на 14 и 20 дни культивирования. На микрофотографиях показаны участки дендритов нейронов. Относительное количество шипиков разных типов отмечено на круговой диаграмме: зеленый — грибовидные шипики, красный — тонкие шпики, черный — тонкие шипики. На 20 день культивирования на поверхности нейронов YAC128 снижается доля грибовидых шипиков и возрастает доля пеньковых. б — Плотность дендритных шипиков (среднее количество шипиков на участке дендрита длиной 10 мкм) на нейронах дикого типа и YAC28.
Поэтому мы сравнили электрофизиологические характеристики двух линий. Это возможно благодаря электрической активности нейронов, которую можно зарегистрировать с помощью специального метода, называемого пэтч-кламп. Для этого к поверхности нейрона прикладывают тонкую стеклянную пипетку, внутри которой находится электрод. Когда нейрон активируется, на его клеточной мембране изменяется напряжение, и это изменение регистрируется электродом. Если взять два связанных синаптическим контактом нейрона и, возбуждая один, регистрировать ответную электрическую активацию на втором, можно измерить эффективность передачи сигнала через синапс (рис. 6). Если ответная активация возникает не всегда, то, вероятно, синаптическая передача ослаблена. Этот метод может выявить нарушения работы синапса до изменений морфологии нейрона.
Рисунок 6. Схема эксперимента при совместном применении оптогенетики и электрофизиологической регистрации. Нейрон коры активируется при освещении синим светом, а на контактирующем с ним нейроне стриатума производится регистрация ответной активности с помощью стеклянной пипетки с электродом.
Чтобы активировать нейрон, можно использовать несколько способов. Можно добавить в окружающую его жидкость химическое вещество, открывающее ионные каналы, что приводит к электрическому возбуждению нейрона. Можно стимулировать нейрон электрическим током. Но мы для своих экспериментов выбрали более тонкий инструмент воздействия на нейроны, а именно — оптогенетику. Этот подход основан на внесении в нейроны специальных светочувствительных белков — опсинов, в результате чего и сами нейроны становятся чувствительными к свету (рис. 7). В результате, освещая нейроны светом определенной длины волны, можно изменять их активность. Освещение синим светом возбуждает нейрон, а желтым — вызывает торможение (т.е. подавляет активность нейрона).
Оптогенетические технологии в наши дни обещают даже вернуть зрение людям с дегенеративными поражениями сетчатки, придав световую чувствительность не разрушенным фоторецепторам, а клеткам-ганглиям: «Оптогенетика + голография = прозрение?» [11]. — Ред.
— Ред.
Рисунок 7. Опсины — светочувствительные белки, обеспечивающие движение ионов через клеточную мембрану и изменение активности нейронов. Каналородопсин (ChR2) вызывает деполяризацию мембраны и активацию нейрона, галородопсин (NpHR) вызывает гиперполяризацию мембраны и торможение нейрона.
Оптогенетика
Оптогенетика — метод, объединяющий подходы генетики и оптики для тонкого контроля электрической активности электровозбудимых клеток (нейронов и мышечных волокон) [9]. Для этого в изучаемые клетки вводят гены специальных светочувствительных белков — микробных опсинов, которые являются ионными каналами или насосами (рис. 7). Первая работа, показавшая возможность управлять электрической активностью нейронов при использовании опсина, была опубликована в 2005 году. За последующие несколько лет появился еще ряд экспериментальных работ, позволивших доработать эту методику и доказать ее применимость в различных экспериментальных условиях.
За последние годы было открыто множество различных опсинов, из которых наибольшее применение в оптогенетике нашли галородопсины и каналородопсины. При доставке гена опсина с помощью методов генной инженерии в нейрон, на плазматической мембране появляются светочувствительные каналы, а сама клетка становится светочувствительной. При действии синего света открывается пора каналородопсина (максимум поглощения — 470 нм), который вызывает движение положительно заряженных ионов внутрь клетки, обеспечивая деполяризацию мембраны нейрона и генерацию потенциалов действия. При действии желтого света активируется галородопсин (максимум поглощения — 580 нм), мембрана нейрона гиперполяризуется, вызывая торможение нейрона. Высокое временное разрешение метода оптогенетики позволяет обеспечить очень тонкую регуляцию синаптических событий и является, таким образом, важным инструментом для изучения межнейронных связей.
Совместное применение оптогенетики и классических методов электрофизиологии позволяет извлечь выгоду из положительных качеств каждого из этих подходов. Точность электрофизиологической регистрации объединяется с возможностью использовать световые стимулы разной длительности и интенсивности, что помогает ученым подробно изучать работу нейронных связей.
Точность электрофизиологической регистрации объединяется с возможностью использовать световые стимулы разной длительности и интенсивности, что помогает ученым подробно изучать работу нейронных связей.
Электрическая активность нейронов выражается в резких скачках напряжения на клеточной мембране и появлении вследствие этого электрического тока. Эти резкие скачки называются спайками или потенциалами действия и длятся несколько миллисекунд (рис. 8). Мы выяснили, что чем дольше нейрон освещается синим светом, тем больше спайков он за это время создает. Если освещаемый нейрон связан синаптическим контактом с другим нейроном, то на втором нейроне можно зарегистрировать ответную активность — тоже в виде отдельных спайков.
Рисунок 8. Пример записи, получаемой при электрофизологической регистрации активности нейрона. Спайки (потенциалы действия) отражаются на записи в виде вертикальных линий, показывающих резкие скачки мембранного тока.
В наших экспериментах мы использовали пару молодых (14 дней) нейронов коры и стриатума, связанных синаптическим контактом. Нейрон коры активировали синим светом, а на нейроне стриатума производили регистрацию ответной активности. Оказалось, что для возникновения ответа на нейроне стриатума нужна определенная длительность освещения (порог активации). Если длительность освещения была ниже порогового значения, то спайк на нейроне стриатума возникал в ответ не на каждую вспышку света. И что самое важное: порог активации для нейронов из мозга здоровых мышей отличался от мышей YAC128 (с мутацией в гене хантингтина). Наиболее ярко эта разница была видна при 50%-активации нейрона стриатума, т.е. длительности освещения, при которой спайк возникает в ответ на каждую вторую вспышку света. Порог 50%-активации нейрона стриатума в ответ на облучение кортикального нейрона для культуры клеток YAC 128 был примерно в два раза (точнее в 2,3±0,8) выше по сравнению с положительным контролем [10].
Нейрон коры активировали синим светом, а на нейроне стриатума производили регистрацию ответной активности. Оказалось, что для возникновения ответа на нейроне стриатума нужна определенная длительность освещения (порог активации). Если длительность освещения была ниже порогового значения, то спайк на нейроне стриатума возникал в ответ не на каждую вспышку света. И что самое важное: порог активации для нейронов из мозга здоровых мышей отличался от мышей YAC128 (с мутацией в гене хантингтина). Наиболее ярко эта разница была видна при 50%-активации нейрона стриатума, т.е. длительности освещения, при которой спайк возникает в ответ на каждую вторую вспышку света. Порог 50%-активации нейрона стриатума в ответ на облучение кортикального нейрона для культуры клеток YAC 128 был примерно в два раза (точнее в 2,3±0,8) выше по сравнению с положительным контролем [10].
Получается, что мутация в белке хантингтине приводит к тому, что синаптическая передача ухудшается уже в молодых нейронах, и при этом нарушения происходят на функциональном уровне (без морфологических изменений). Может ли быть так, что именно эти функциональные нарушения в дальнейшем приводят к появлению морфологических изменений, таких как исчезновение шипиков у старых нейронов стриатума?
Может ли быть так, что именно эти функциональные нарушения в дальнейшем приводят к появлению морфологических изменений, таких как исчезновение шипиков у старых нейронов стриатума?
Чтобы ответить на этот вопрос, мы снова обратились за помощью к оптогенетике, но теперь с ее помощью мы подавляли светом активность нейронов (рис. 9а). Если наше предположение верно, то временное отсутствие активирующего воздействия коры должно привести к исчезновению шипиков на нейронах стриатума в культуре YAC128. Действительно, после эксперимента количество шипиков в положительном контроле осталось неизменным, но в культуре YAC128 значительно снизилось (рис. 9б, в). Получается, что моделирующие БХ нейроны особенно чувствительны к ослаблению активирующего воздействия нейронов коры, поэтому длительное ослабление синаптической связи между этими нейронами приводит к уменьшению количества шипиков на 20 день культивирования [10].
Рисунок 9. Влияние на нейроны с помощью оптогенетики. а — подавление активности нейрона коры с помощью оптогенетики. При освещении нейрона желтым светом (оранжевая полоса) он становится неактивен — на электрофизиологической записи в этот промежуток времени почти нет спайков. б — влияние длительного оптогенетического торможения на морфологию нейронов. На микрофотографиях видно уменьшение количества шипиков на дендрите нейрона YAC128. в — изменение плотности дендритных шипиков после оптогенетического торможения. В культуре дикого типа количество шипиков не изменяется, а в культуре YAC128 плотность шипиков значительно снижается.
а — подавление активности нейрона коры с помощью оптогенетики. При освещении нейрона желтым светом (оранжевая полоса) он становится неактивен — на электрофизиологической записи в этот промежуток времени почти нет спайков. б — влияние длительного оптогенетического торможения на морфологию нейронов. На микрофотографиях видно уменьшение количества шипиков на дендрите нейрона YAC128. в — изменение плотности дендритных шипиков после оптогенетического торможения. В культуре дикого типа количество шипиков не изменяется, а в культуре YAC128 плотность шипиков значительно снижается.
Таким образом, мы обнаружили, что в нашей модели БХ наблюдается нарушение синаптической передачи, которое развивается в два этапа. На ранней стадии (молодые нейроны, на 14 день культивирования нейронов) происходит функциональное ослабление сипаптической связи, которое на более поздней стадии (старые нейроны, на 20 день культивирования) приводит к морфологическим нарушениям синаптических контактов.
Вывод: нарушение синаптической передачи и нейродегенеративные заболевания
Исследование подтвердило нашу первоначальную догадку: нарушение работы стриатума при БХ связано, прежде всего, с нарушением синаптической передачи. Нейроны стриатума перестают «слышать» команды нейронов коры, и, в результате, человек начинает терять контроль над своими движениями. Но на ранней стадии заболевания эти нарушения носят функциональный характер и, вероятно, обратимы. Если выяснить причины, ведущие к нарушению работы синапсов, то возможно разработать лекарственное средство, которое поможет остановить патологические изменения, обеспечит нейронам стриатума необходимый уровень активации и, таким образом, предотвратит развитие необратимых морфологических нарушений.
Результаты этой работы подталкивают и к другому важному заключению: при изучении нейродегенеративных заболеваний ученым следует обратить больше внимания на синаптическую передачу и взаимодействие нейронов. Мы знаем достаточно много о том, как влияет накопление амилоидных агрегатов при болезни Альцгеймера на жизнедеятельность клетки, и какие нейроны погибают при развитии болезни Паркинсона, но это все еще не привело к появлению эффективной терапии. Возможная причина этих неудач — мы не учитываем последствия нарушения синаптических связей — те, которые мы открыли при исследовании болезни Хантингтона. Хочется надеяться, что наши находки подскажут исследователям направление для разработки эффективных способов лечения болезни Хантингтона, и Тринадцатая наконец-то будет здоровой!
Возможная причина этих неудач — мы не учитываем последствия нарушения синаптических связей — те, которые мы открыли при исследовании болезни Хантингтона. Хочется надеяться, что наши находки подскажут исследователям направление для разработки эффективных способов лечения болезни Хантингтона, и Тринадцатая наконец-то будет здоровой!
Исследование было проведено в лаборатории молекулярной нейродегенерации СПбГПУ (зав. лаб. — проф. Техасского университета Илья Борисович Безпрозванный) под руководством к.б.н. Дмитрия Николаевича Артамонова.
- Hayden M.R. Huntington’s Chorea. New York: Springer, 1982;
- DR Langbehn, RR Brinkman, D Falush, JS Paulsen, MR Hayden, on behalf of an International Huntington’s Disease Collaborative Group. (2004). A new model for prediction of the age of onset and penetrance for Huntington’s disease based on CAG length. Clinical Genetics. 65, 267-277;
- Идентифицированы белки, «слипающиеся» при болезни Гентингтона;
- Sara Imarisio, Jenny Carmichael, Viktor Korolchuk, Chien-Wen Chen, Shinji Saiki, et. al.. (2008). Huntington’s disease: from pathology and genetics to potential therapies. Biochem. J.. 412, 191-209;
- Claire-Anne Gutekunst, Shi-Hua Li, Hong Yi, James S. Mulroy, Stefan Kuemmerle, et. al.. (1999). Nuclear and Neuropil Aggregates in Huntington’s Disease: Relationship to Neuropathology. J. Neurosci.. 19, 2522-2534;
- Boxun Lu, James Palacino. (2013). A novel human embryonic stem cell-derived Huntington’s disease neuronal model exhibits mutant huntingtin (mHTT) aggregates and soluble mHTT-dependent neurodegeneration. The FASEB Journal. 27, 1820-1829;
- Benjamin Ray Miller, Ilya Bezprozvanny. (2010). Corticostriatal circuit dysfunction in Huntington’s disease: intersection of glutamate, dopamine and calcium. Future Neurology. 5, 735-756;
- Нобелевская премия по физиологии и медицине (2013): везикулярный транспорт;
- Karl Deisseroth. (2011). Optogenetics. Nat Methods. 8, 26-29;
- Артамонов Д.Н., Коржова В.В., Ву Д., Рыбальченко П.Д., Им К., Красноборова В.А., Власова О.Л., Безпрозванный И.Б. (2013). Нарушения синаптической передачи при болезни Хантингтона на кортикостриатной модели культуры нейронов. «Биологические мембраны». 30, 1–13;
- Оптогенетика + голография = прозрение?.
 al.. (2008). Huntington’s disease: from pathology and genetics to potential therapies. Biochem. J.. 412, 191-209;
al.. (2008). Huntington’s disease: from pathology and genetics to potential therapies. Biochem. J.. 412, 191-209; (2011). Optogenetics. Nat Methods. 8, 26-29;
(2011). Optogenetics. Nat Methods. 8, 26-29;История создания и развития лабораторий Гемохелп
Рейтинг статьи: 4,56 (264)
| История и философия ГЕМОХЕЛП | |
| Лабораторное оборудование | |
| Лицензии | |
| Контроль качества | |
| Специалисты | |
| Сотрудничество с ФГБОУ ВО «ПИМУ» | |
| «ГЕМОХЕЛП» — участник Ассоциации нижегородских частных медицинских центров | |
| Наши партнеры | |
| Контакты | |
| Новости |
В 2013 году, накануне своего восьмилетия, компания прошла процедуру ребрендинга и получила название «ГЕМОХЕЛП» и главное в этом слове «ХЕЛП», т. е. «ПОМОЩЬ», помощь вам Быть Здоровыми. Все началось в 2005 г, когда однажды успешный нижегородский предприниматель-инвестор Иванушкин Сергей Александрович, будучи в гостях у своих родителей, застал подругу мамы в крайне расстроенном состоянии. Женщина объяснила, что вернулась из медицинского учреждения, где выстояла очередь, но так и не смогла взять талончик на анализ, талонов всем желающим не хватило. Назавтра предстояла та же неприятная процедура и неизвестный результат.
е. «ПОМОЩЬ», помощь вам Быть Здоровыми. Все началось в 2005 г, когда однажды успешный нижегородский предприниматель-инвестор Иванушкин Сергей Александрович, будучи в гостях у своих родителей, застал подругу мамы в крайне расстроенном состоянии. Женщина объяснила, что вернулась из медицинского учреждения, где выстояла очередь, но так и не смогла взять талончик на анализ, талонов всем желающим не хватило. Назавтра предстояла та же неприятная процедура и неизвестный результат.
Так возникла идея создания независимой централизованной лаборатории, которая открыла свои двери уже в январе 2006 года. Для удобства клиентов и приближения лабораторных улуг к дому стали открываться процедурные кабинеты в разных районах города.
После открытия трех процедурных кабинетов, которые находились на территории поликлиник, в марте 2006 года открылся первый фирменный кабинет на улице Горького.
Сегодня компания «ГЕМОХЕЛП» – Лидер рынка медицинских анализов в Нижнем Новгороде и Нижегородской области. Это группа компаний, которая включает централизованную лабораторию, оборудованную по европейским стандартам и 74 процедурных кабинетов в городах: Нижний Новгород, Дзержинск, Бор, Арзамас, Балахна, Богородск, Кстово, Выкса, Чебоксары, Павлово, Муром, Меленки, Заволжье, Лысково, Саров, Лукоянов, Вача, Кулебаки, Навашино, Семенов, Сосновское, Чкаловск. Высокопрофессиональный штат специалистов, ежедневное обслуживание более 2000 человек и ежедневное выполнение порядка 20 000 исследований.
Это группа компаний, которая включает централизованную лабораторию, оборудованную по европейским стандартам и 74 процедурных кабинетов в городах: Нижний Новгород, Дзержинск, Бор, Арзамас, Балахна, Богородск, Кстово, Выкса, Чебоксары, Павлово, Муром, Меленки, Заволжье, Лысково, Саров, Лукоянов, Вача, Кулебаки, Навашино, Семенов, Сосновское, Чкаловск. Высокопрофессиональный штат специалистов, ежедневное обслуживание более 2000 человек и ежедневное выполнение порядка 20 000 исследований.
«ГЕМОХЕЛП» активно сотрудничает с более чем 200 частными и муниципальными лечебными учреждениями, более чем с 2500 практикующими врачами.
«Жизнь есть Любовь! Наша задача открыться довериться энергиям Любви и радостно путешествовать по ее просторам!!! И когда это случается – ее крылья легко несут тебя по дороге Жизни.
Бизнес — одна из составных граней Жизни! Поэтому для меня естественно строить мои предприятия на фундаменте Любви и наполнять их добротой, состраданием, чистотой и гармонией!!! Любовь, когда вы чисты, протекает через вас, превращая Жизнь окружающих в Праздник!
Лаборатория «Гемохелп» рождена из желания помочь гражданам не стоять в очередях за талончиками в государственных лечебных учреждениях и получать объективные, независимые анализы для оценки своего здоровья. Без соучастия, сострадания, желания помочь – в медицине делать нечего.
Без соучастия, сострадания, желания помочь – в медицине делать нечего.
Я развиваю «Гемохелп». Для меня это история, построенная именно на любви- любви к своим сотрудникам, к своим клиентам, партнерам. Я считаю, что если бизнес несет любовь, то он будет расти. А прибыль для меня никогда не была целью, это лишь результат, аплодисменты рынка.
Вся работа компании нацелена на служение и помощь нашим клиентам. Мы делаем это с радостью! Новейшее оборудование, обеспечивающие достоверные и точные результаты исследований, просторные, светлые процедурные кабинеты в каждом районе Нижнего Новгорода и в районных центрах, профессиональный, заботливый персонал, минимальные сроки выполнения большинства исследований (1 рабочий день) – это выражение нашей заботы и любви к клиентам. Обратившись в «Гемохелп», любой человек может рассчитывать на грамотные советы по вопросам, связанными с лабораторными исследованиями. Мы искренне стремимся к тому, чтобы обращение к нам было комфортным и максимально приятным опытом общения с медицинской лабораторией.
Мы любим своих партнеров, ценим и прислушиваемся к их мнению, стараемся индивидуально для каждого создать наиболее выгодные условия для сотрудничества.
Вся жизнь базируется на энергиях любви, и каждый человек – творец своего счастья.
Пусть Любовь наших сердец, наша доброта и теплота, сострадание и профессионализм помогает Вам Быть Здоровыми и Радостными!!!»
С любовью Сергей Иванушкин.
Иванушкин Сергей Александрович родился 23 апреля 1958 года в городе Горьком.
Учился в физико-математической школе №40. Окончил Горьковский Государственный Университет им. Н.И.Лобачевского, экономический факультет, в 2001 году — Академию Народного Хозяйства при Правительстве Российской Федерации по специальности «Современный руководитель производства». Защитил диссертацию и удостоился ученой степени кандидата экономических наук.
С 1992 года является собственником бизнеса. Занимался строительством кораблей, открывал химчистки, call-центры, рестораны.
В 2003 году создал частный семейный Благотворительный фонд «Начало» семьи Иванушкиных. Основное направление фонда — помощь детям, воспитывающимся в детских домах и школах-интернатах, детям-инвалидам, детям, оставшимся без родителей, перспективным и одаренным детям.
В июле 2006 года по инициативе Иванушкина С.А. началась реализация первого в России социального проекта Банк Времени. Фонд оказывает материальную, техническую и гуманитарную помощь в рамках собственных благотворительных программ.
За время работы частный семейный Благотворительный Фонд «Начало» удостоен почетного Диплома «Фирма Доброй воли 2003 года», в 2004 году Иванушкин С.А. стал победителем в номинации «Благотворитель года» и «Меценат года» — в 2005 году. В 2014 году награжден золотой медалью «Благотворитель земли Нижегородской», а 08 декабря 2014 г. Сергей Александрович Иванушкин стал победителем премии «Человек года» в сфере медицинских технологий.
С 2003 года учредитель, президент группы компаний AVK.
Женат. Дети: три дочери, сын, два внука и три внучки.
Используя сайт gemohelp.ru, вы соглашаетесь с использованием файлов cookie
Подтверждаю
Подробнее
Болезнь Гентингтона — Вики
Болезнь Гентингтона (синдром Гентингтона, хорея Гентингтона, хорея Хантингтона[3]) — аутосомно-доминантное генетическое заболевание нервной системы, характеризующееся постепенным началом обычно в возрасте 30—50 лет и сочетанием прогрессирующего хореического гиперкинеза и психических расстройств. Заболевание вызывается умножением кодона CAG в гене HTT. Этот ген кодирует 350-kDa белок гентингтин с неизвестной функцией. В гене дикого типа (не мутантного) у разных людей присутствует разное количество CAG-повторов, однако, когда число повторов превышает 36, развивается болезнь. Нейроморфологическая картина характеризуется атрофией стриатумa, а на поздней стадии также атрофией коры головного мозга.
Содержание
- 1 Эпидемиология
- 2 Генетика
- 3 Патогенез
- 3. 1 Функция Htt
- 3.2 Клеточные изменения под действием mHtt
- 3.3 Макроскопические изменения под действием mHtt
- 3.
- 4 Симптоматика
- 5 Диагностика
- 5.1 Клинические методы
- 5.2 Генетические методы
- 5.3 Эмбрионы
- 5.4 Дифференциальная диагностика
- 6 Лечение
- 7 Прогноз
- 8 Направление исследований
- 8.1 Обзор по клиническим исследованиям
- 8.2 Снижение уровня гентингтина
- 9 Примечания
- 9.1 Комментарии
- 9.2 Источники
- 10 Литература
 1 Функция Htt
1 Функция HttЭпидемиология
В настоящее время от хореи Гентингтона в США страдает около 7000 человек. Частота встречаемости заболевания среди населения с европейскими корнями составляет примерно 3–7:100000 и 1:1000000 среди остальных рас[4][нет в источнике]. Название болезни дано в честь трёх поколений врачей, изучавших её в штате Коннектикут. В частности, считается, что заболевание названо в честь американского врача Джорджа Хантингтона (Гентингтона), первым давшего его классическое описание[5][6][a].
Генетика
Ген HTT, присутствующий у всех людей, кодирует белок гентингтин (Htt). Ген HTT расположен на коротком плече 4-й хромосомы (4p16.3)[7]. Этот ген содержит в себе участок с повторяющейся последовательностью трёх азотистых оснований — цитозин-аденин-гуанин (то есть, ЦАГЦАГЦАГ…). Триплет ЦАГ кодирует аминокислоту глутамин, поэтому синтезируемый белок гентингтин содержит последовательность глутаминовых аминокислот, называемую полиглутаминовый тракт[8].
Количество ЦАГ триплетов различно у отдельных лиц и может изменяться с последующими поколениями. Если их становится больше 36, то синтезируется удлинённый полиглутаминовый тракт и происходит образование мутантного белка гентингтина (mHtt)[9], который оказывает токсичное действие на клетки и вызывает болезнь Гентингтона. Как правило, от числа ЦАГ повторов зависит степень повреждений, наличие около 60 % повторов сверх нормы вызывает появление симптомов в различном возрасте[7]. 36—40 повторов приводят к редуцированной пенетрантности формы этого заболевания, которая намного позже проявляется и медленнее прогрессирует. В некоторых случаях начало болезни может быть настолько поздним, что симптомы никогда не обнаруживаются[10]. При очень большом количестве повторов болезнь Гентингтона имеет полную пенетрантность и может проявиться до 20 лет, тогда болезнь классифицируется как ювенильная, акинетически-ригидная или Вестфаль варианты. Составляет приблизительно 7 % случаев болезни Гентингтона[11].
Мутантный ген был предположительно завезён в США в 1630 году двумя братьями, эмигрировавшими из Эссекса в Бостон[12][13].
Болезнь передаётся по наследству. Мутантный аллель доминантный, поэтому в семье, где один из родителей несёт такую мутацию, каждый из потомков может получить её с вероятностью 50 %. Наследование не зависит от пола носителя или его детей.
Патогенез
Htt-белок взаимодействует с сотней других белков и, вероятно, выполняет множество биологических функций[14]. Механизм действия mHtt до конца не ясен, но известно, что он токсичен для некоторых типов клеток, особенно в головном мозге. В основном происходит поражение полосатого тела (стриатума), но при прогрессировании заболевания и другие области головного мозга значительно повреждаются[9]. Планирование и коррекция движений — основная функция полосатого тела, и нарушения в этой области провоцируют симптомы[9].
Функция Htt
Htt образуется во всех клетках млекопитающих. Наибольшая его концентрация — в головном мозге и яичках, а также в умеренных количествах в печени, сердце и лёгких[9]. Функция Htt у человека не ясна. Он взаимодействует с белками, участвующими в транскрипции, передаче сигнала в клетке и внутриклеточном транспорте[9][15]. Некоторые функции Htt обнаружены в экспериментальных моделях животных: играет важную роль в развитии эмбриона и связан с гибелью эмбриона при отсутствии белка[16]. Он также выступает в качестве анти-апоптозного агента, предотвращая запрограммированную гибель клеток, и контролирует образование нейротрофического фактора мозга (белок, защищающий нейроны и регулирующий их образование во время нейрогенеза). Если экспрессия Htt возрастает, выживаемость нервных клеток увеличивается и эффект mHtt уменьшается, наоборот, понижение экспрессии Htt даёт картину более типичную присутствию mHtt[16]. У людей разрушение нормального гена не приводит к болезни. В настоящее время считается, что болезнь вызывает не недостаточное образование Htt, а усиление токсического эффекта mHtt[9].
Клеточные изменения под действием mHtt
Под действием образовавшегося mHtt происходит множество изменений в клетке, что вызывает болезнь Гентингтона. Удлинение полиглутаминовой последовательности изменяет конформацию белка гентингтина и прочно соединяет его с другими белками[17]. Это приводит к агрегации гентингтина, при этом образуются так называемые внутриклеточные тельца включения[18]. Эти включения механически препятствуют движению везикул, содержащих нейромедиаторы, через цитоскелет, что нарушает передачу сигналов в нейронах[18]. Тельца включения обнаруживаются как в ядрах клеток, так и в цитоплазме. Некоторые эксперименты показали, что они могут быть токсичны для клеток, а другие — что тельца, наоборот, защищают нейрон от смерти, аккумулируя мутантный гентингтин, и именно неагрегированный белок токсичен[19].
Существует несколько путей, при которых mHtt вызывает гибель клеток. К ним относят: влияние на белки-шапероны; взаимодействие с каспазами, которые участвуют в апоптозе; токсическое действие глутамина на нервные клетки; нарушение выработки энергии в клетках и влияние на экспрессию генов. Токсическое действие mHtt значительно усиливается при взаимодействии с белком RASD2 (Rhes), который образуется преимущественно в стриатуме. RASD2 вызывает сумоляцию (SUMOylation) mHtt к образованию белковых сгустков и дезагрегации — исследования в культуре клеток показали, что сгустки менее токсичны, чем дезагрегированная форма[20].
Макроскопические изменения под действием mHtt
Область головного мозга, поражающаяся при болезни Гентингтона, — стриатум (выделена малиновым цветом)
Болезнь Гентингтона поражает специфические области мозга. Наиболее заметные ранние изменения затрагивают область базальных ганглиев, называемую полосатым телом, которое состоит из хвостатого ядра и скорлупы[9]. Другие повреждаемые области включают чёрную субстанцию, 3, 5 и 6 слои коры головного мозга, гиппокамп, клетки Пуркинье в мозжечке, боковые туберальные ядра гипоталамуса и часть таламуса[7]. Эти области получают повреждения в соответствии с их структурой и типами содержащихся в них нейронов, уменьшаясь в размерах в связи с гибелью клеток[7]. Звёздчатые нейроны полосатого тела наиболее уязвимы, особенно проецирующиеся в направлении поверхности бледного шара, вставочные и звёздчатые нейроны, проецирующиеся к центру бледного шара, получают меньше повреждений[7][21]. Болезнь Гентингтона также вызывает аномальное увеличение астроцитов[22].
Базальные ганглии — часть головного мозга, наиболее заметно повреждающиеся при болезни Гентингтона — играют ключевую роль в контроле движений и поведения. Их функция полностью не ясна, но современные теории предполагают, что они являются частью когнитивной исполнительной системы.
Базальные ганглии в норме ингибируют большое число контуров (circuit), генерирующих специфические движения. Для инициации специфических движений кора посылает сигналы базальным ганглиям для снятия ингибирования. Повреждение базальных ганглиев может привести к снятию ингибирования или его постоянным неконтролируемым изменениям, что служит причиной затруднения начала движения или к их непроизвольной инициации, или движение может быть прервано до или после достижения желаемого результата. Накапливающиеся повреждения в этой области приводят к беспорядочным движениям, характерным для болезни Гентингтона[23].
Симптоматика
Симптомы болезни Гентингтона могут проявиться в любом возрасте, но чаще это происходит в 35—44 года[24][25]. На ранних стадиях происходят небольшие изменения личности, когнитивных способностей и физических навыков[24]. Обычно первыми обнаруживают физические симптомы, так как когнитивные и психические расстройства не столь выражены в ранних стадиях[24]. Почти у всех пациентов болезнь Гентингтона в итоге проявляется схожими физическими симптомами, но начало заболевания, прогрессирование и степень когнитивных и психических нарушений различаются у отдельных лиц[26][27].
Для начала заболевания наиболее характерна хорея — беспорядочные, неконтролируемые движения. Хорея вначале может проявляться в беспокойстве, небольших непроизвольных или незавершённых движениях, нарушении координации и замедлении скачкообразных движений глаз[24].
В самом начале обычно возникают проблемы из-за физических симптомов, которые выражаются в резких, внезапных и не поддающихся контролю движениях. В других случаях, наоборот, больной двигается слишком замедленно. Возникают нарушения координации движений, речь становится невнятной. Постепенно все функции, требующие мышечного контроля, нарушаются: человек начинает гримасничать, испытывает проблемы с жеванием и глотанием. Из-за быстрого движения глаз происходят нарушения сна. Обычно больной проходит через все стадии физического расстройства, однако влияние болезни на когнитивные функции у всех очень индивидуально. Чаще всего происходит расстройство абстрактного мышления, человек перестаёт быть способным планировать свои действия, следовать правилам, оценивать адекватность своих действий. Постепенно появляются проблемы с памятью, могут возникнуть депрессия и паника, эмоциональный дефицит, эгоцентризм, агрессия, навязчивые идеи, проблемы с узнаванием других людей, гиперсексуальность и усиление вредных привычек, таких как алкоголизм или игромания. Бывает, что человек, болеющий хореей Гентингтона, не понимает, сыт он или голоден. Тогда больных приходится кормить 3—4 раза в день и не перекармливать.
Диагностика
Клинические методы
Физикальное обследование, иногда в сочетании с психологическим обследованием, позволяет определить область распространения болезни[24]. Медицинская визуализация (компьютерная томография (КТ), магнитно-резонансная томография (МРТ)) показывает только видимую атрофию мозга на прогрессирующей стадии заболевания. Методы функциональной нейровизуализации (фМРТ и позитронно-эмиссионная томография (ПЭТ)) могут показать изменения в активности мозга до появления клинических симптомов[7].
Генетические методы
Для проведения генетической диагностики болезни Гентингтона необходим забор крови с последующим определением количества повторов ЦАГ в каждом НТТ-аллеле[28]. Положительный результат не подтверждает диагноз, поскольку может быть получен за несколько лет до появления первых симптомов. Однако отрицательный результат однозначно свидетельствует об отсутствии вероятности развития болезни Гентингтона[7].
Эмбрионы
Эмбрионы, полученные в результате экстракорпорального оплодотворения, могут быть подвержены генетической диагностике болезни Гентингтона с применением преимплантационной генетической диагностики. При этом методе забирается одна клетка из 4-8-клеточного эмбриона и затем проверяется на генетическую патологию. Полученная информация может впоследствии быть использована при выборе здорового эмбриона для имплантации. Кроме того, возможна пренатальная диагностика для эмбриона или плода в утробе матери[29].
Дифференциальная диагностика
Около 90 % диагнозов болезни Гентингтона, основанных на обнаружении типичных симптомов и семейном анамнезе, подтверждаются генетическим тестированием. Большинство других расстройств с аналогичными симптомами называют ХГ-подобными расстройствами (англ. HD-like disorders, HDL)[30]. Причины большинства HDL-заболеваний неизвестны. Известно лишь, что некоторые из них возникают в результате мутаций генов PRNP (HDL1), junctophilin 3 (HDL2), рецессивно наследуемого HTT гена (HDL3 — обнаружен у одной семьи и мало изучен) и гена, кодирующего ТАТА-связывающий белок (HDL4/SCA17)[30]. К другим заболеваниям с аутосомно-доминантным наследованием, которые схожи с болезнью Гентингтона, относят дентаторубро-паллидолюисовую атрофию и нейроферритинопатию[30].
Лечение
Химическая структура тетрабеназина, разрешённого для лечения болезни Гентингтона
Болезнь Гентингтона неизлечима, но существует лечение, способное облегчить некоторые симптомы. [31]
Тетрабеназин был разработан специально для уменьшения тяжести симптомов болезни Гентингтона,[32] был утвержден в 2008 году в США.[33]Нейролептики и бензодиазепины помогают уменьшить проявления хореи[25]. Амантадин и ремацемид находятся в стадии исследования, но показали положительные результаты.[34] Для облегчения гипокинезии и ригидности мышц назначают противопаркинсонические лекарства, для облегчения миоклонической гиперкинезии — вальпроевую кислоту.[35] В России препарат продается под торговым названием Нормокинезтин. С 1 января 2018 года Нормокинезтин включен в обновленный перечень жизненно необходимых лекарственных препаратов.
Для устранения депрессии применяют селективные ингибиторы обратного захвата серотонина и миртазапин, а при психозах и нарушениях поведения назначают атипичные антипсихотики[36].
В настоящий момент ведутся активные исследования по разработке способа лечения, исследуются потенциальные направления для лечения болезни Гентингтона[37]. Так компания Teva исследовала препарат-иммуномодулятор лахинимод, обладающий протективным действием по отношению к ЦНС. Испытания препарата дошли до II фазы, но в ходе КИ препарату не удалось достигнуть конечной точки оценки эффективности. Однако испытующие определили, что на фоне терапии лахинимодом происходит снижение скорости атрофии головного мозга. Исходя из опыта неудавшегося исследования, компания приняла решение об отказе от дальнейшего изучения лекарственного препарата[38][39].
Прогноз
С момента появления первых симптомов продолжительность жизни составляет около 15—20 лет.
Смерть обычно происходит не из-за болезни Гентингтона, а из-за сопутствующих ей осложнений, включая пневмонию, заболевания сердца и травмы. Часто больные совершают суицид.
Направление исследований
Обзор по клиническим исследованиям
На ноябрь 2021 года по всему миру было запущено 208 клинических исследований[40], 59 из них находятся в активном статусе[41].
Клинические исследования используют различные методики лечения.
Снижение уровня гентингтина
Причиной БГ является наличие мутации в одном гене, который вырабатывает токсичный для клеток вариант белка гентингтина. Одним из самых перспективных способов лечения исходит из идеи снизить уровень выработки этого варианта белка путём использования технологий подавления экспрессии генов. Исследования на мышиных моделях указывают на то, что снижение уровня мутантного белка в нервной системе улучшает их состояние [42]. Безопасность некоторых методов подавления экспрессии генов, например, РНК-интерференции или использования аллель-специфичных олигонуклеотидов (ASO), проверена в исследованиях на мышиных моделях, а также в мозге макак[43][44]. У аллель-специфичного подавления имеется возможность уменьшать количество как обоих вариантов белка (дикого и мутантного), так и быть специфичным только к мутантному, например, с помощью определения однонуклеотидного полиморфизма между двумя вариантами гена[45]. В качестве механизма подавления рассматриваются и технологии редактирования генома, например, с помощью систем на базе CRISPR/Cas9[46].
По ASO уже имеются определённые результаты по нескольким исследованиям. В 2015 году компания Ionis Pharmaceuticals совместно и Институтом Нейрологии UCL запустили Фазу 1/2a клинического исследования препарата IONIS-HTTRx. Данный этап исследования должны был показать как безопасность использования на людях, так и получить первую информацию об эффективности. IONIS-HTTRx призван уменьшать экспрессию обеих копий гена. В качестве метода обнаружения снижения уровня белка гентингтина был взят метод измерения его содержания в спинномозговой жидкости. Результаты этой Фазы показали, что у всех участников, кому вводили препарат, обнаруживается статистически значимое снижения уровня гентингина. Фаза 3 была начата в 2019 году при партнёрстве с Roche Pharmaceuticals. Препарат к этому моменту был переименован на «томинерсен». Однако в марте 2021 года исследование томинерсена было остановлено регулирующими организациями из-за выявления неоптимального соотношения пользы и риска.
Имеются другие исследования, которые исследуют возможности подавления эспрессии генов. Компания Wave Life Sciences в 2021 году закончила клиническое тестирование двух препаратов, которые специфичны только для мутантного варианта гена. Результаты исследования этих препаратов оказались неудачными, на данный момент Wave Life Sciences проводит подготовку для начала тестирования третьего варианта препарата. Компания Uniqure в 2019 году анонсировала свой вариант ASO.
Примечания
Комментарии
- ↑ Джордж Хантингтон, описавший в 1872 г болезнь, названную в его честь, был не первым, кто дал ее описание. Это было сделано различными авторами, как минимум пяти публикаций, начиная с 1832 года[3]
Источники
- ↑ база данных Disease ontology (англ.) — 2016.
- ↑ Monarch Disease Ontology release 2018-06-29sonu — 2018-06-29 — 2018.
- ↑ 1 2 Кёрк, 2022, с. 74.
- ↑ NCBI OMIM. Huntington’s Disease (неопр.). Дата обращения: 22 мая 2008. Архивировано 19 ноября 2004 года.
- ↑ George Huntington (1850—1916) and Hereditary Chorea (неопр.). Дата обращения: 29 сентября 2011. Архивировано 8 марта 2021 года.
- ↑ George Huntington (1850—1916) and hereditary chorea (неопр.). Дата обращения: 3 октября 2017. Архивировано 19 октября 2017 года.
- ↑ 1 2 3 4 5 6 7 Walker F.O. Huntington’s disease (англ.) // The Lancet. — Elsevier, 2007. — Vol. 369, no. 9557. — P. 220. — doi:10.1016/S0140-6736(07)60111-1. — PMID 17240289.
- ↑ Katsuno M., Banno H., Suzuki K., et al. Molecular genetics and biomarkers of polyglutamine diseases (англ.) // Current Molecular Medicine (англ.) (рус. : journal. — 2008. — Vol. 8, no. 3. — P. 221—234. — doi:10.2174/156652408784221298. — PMID 18473821. Архивировано 13 февраля 2009 года.
- ↑ 1 2 3 4 5 6 7 Walker F.O. Huntington’s disease (англ.) // The Lancet. — Elsevier, 2007. — Vol. 369, no. 9557. — P. 221. — doi:10.1016/S0140-6736(07)60111-1. — PMID 17240289.
- ↑ Walker F.O. Huntington’s disease (англ.) // The Lancet. — Elsevier, 2007. — Vol. 369, no. 9557. — P. 222. — doi:10.1016/S0140-6736(07)60111-1. — PMID 17240289.
- ↑ Nance M.A., Myers R.H. Juvenile onset Huntington’s disease—clinical and research perspectives (англ.) // Developmental Disabilities Research Reviews (англ.) (рус. : journal. — 2001. — Vol. 7, no. 3. — P. 153—157. — doi:10.1002/mrdd.1022. — PMID 11553930.
- ↑ Vessie, P. R. (1932) «On the transmission of Huntington’s chorea for 300 years—the Bures family group». Journal of Nervous and Mental Disease, Baltimore 76: 553—573.
- ↑ Wexler, A. (2008) «The Woman Who Walked into the Sea: Huntington’s and the Making of a Genetic Disease», Yale University Press; 1 edition
- ↑ Goehler H., Lalowski M., Stelzl U., et al. A protein interaction network links GIT1, an enhancer of Huntingtin aggregation, to Huntington’s disease (англ.) // Molecular Cell (англ. ) (рус. : journal. — 2004. — Vol. 15, no. 6. — P. 853—865. — doi:10.1016/j.molcel.2004.09.016. — PMID 15383276.
- ↑ Harjes P., Wanker E.E. The hunt for huntingtin function: interaction partners tell many different stories (англ.) // Trends (англ.) (рус. : journal. — 2003. — Vol. 28, no. 8. — P. 425—433. — doi:10.1016/S0968-0004(03)00168-3. — PMID 12932731.
- ↑ 1 2 Cattaneo E., Zuccato C., Tartari M. Normal huntingtin function: an alternative approach to Huntington’s disease (англ.) // Nature Reviews Neuroscience : journal. — 2005. — Vol. 6, no. 12. — P. 919—930. — doi:10.1038/nrn1806. — PMID 16288298.
- ↑ Rubinsztein D.C., Carmichael J. Huntington’s disease: Molecular basis of neurodegeneration (англ.) // Expert Rev Mol Med : journal. — 2003. — Vol. 5, no. 20. — P. 1—21. — doi:10.1017/S1462399403006549. — PMID 14585171.
- ↑ 1 2 Huntingtin Protein and Protein Aggregation | HOPES — A guide to the science of Huntington’s disease (неопр.) (недоступная ссылка). Архивировано 31 августа 2010 года.
- ↑ Arrasate, M. (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature, 431(7010):805-10.
- ↑ Subramaniam S., Sixt K.M., Barrow R., Snyder S.H. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity (англ.) // Science : journal. — 2009. — Vol. 324, no. 5932. — P. 1327—1330. — doi:10.1126/science.1172871. — PMID 19498170.
- ↑ Purves D., Augustine G. A., Fitzpatrick D., Hall W., LaMantia A-S, McNamara J. O., Williams S. M. Modulation of Movement by the Basal Ganglia – Circuits within the Basal Ganglia System // Neuroscience (англ. ) / Purves D.. — 2nd. — Sunderland, MA: Sinauer Associates (англ.) (рус., 2001. — ISBN 0-87893-742-0.
- ↑ Lobsiger C.S., Cleveland D.W. Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease (англ.) // Nature Neuroscience. — 2007. — Vol. 10, no. 11. — P. 1355—1360. — doi:10.1038/nn1988. — PMID 17965655.
- ↑ Crossman A.R. Functional anatomy of movement disorders (англ.) // Journal of Anatomy (англ.) (рус.. — 2000. — Vol. 196 (Pt 4). — P. 519—525. — doi:10.1046/j.1469-7580.2000.19640519.x. — PMID 10923984. (недоступная ссылка)
- ↑ 1 2 3 4 5 Walker F. O. Huntington’s disease (англ. ) // The Lancet. — Elsevier, 2007. — Vol. 369, no. 9557. — P. 218. — doi:10.1016/S0140-6736(07)60111-1. — PMID 17240289.
- ↑ 1 2 Huntington Disease (неопр.). genereviews bookshelf. University of Washington (19 июля 2007). Дата обращения: 12 марта 2009. Архивировано 10 февраля 2009 года.
- ↑ Kremer B. Clinical neurology of Huntington’s disease // Huntington’s Disease – Third Edition (англ.) / Bates G., Harper P., Jones L.. — Oxford: Oxford University Press, 2002. — P. 28—53. — ISBN 0-19-851060-8.
- ↑ Wagle A. C.; Wagle S. A., Marková I. S., Berrios G. E. Psychiatric Morbidity in Huntington’s disease (англ.) // Neurology, Psychiatry and Brain Research. — 2000. — No. 8. — P. 5—16.
- ↑ Myers R.H. Huntington’s disease genetics (англ. ) // NeuroRx. — 2004. — Vol. 1, no. 2. — P. 255—262. — doi:10.1602/neurorx.1.2.255. — PMID 15717026.
- ↑ Kuliev A., Verlinsky Y. Preimplantation diagnosis: A realistic option for assisted reproduction and genetic practice (англ.) // Curr. Opin. Obstet. Gynecol. : journal. — 2005. — Vol. 17, no. 2. — P. 179—183. — doi:10.1097/01.gco.0000162189.76349.c5. — PMID 15758612.
- ↑ 1 2 3 Schneider S.A., Walker R.H., Bhatia K.P. The Huntington’s disease-like syndromes: what to consider in patients with a negative Huntington’s disease gene test (англ.) // Nature Reviews Neurology : journal. — 2007. — Vol. 3, no. 9. — P. 517—525. — doi:10.1038/ncpneuro0606. — PMID 17805246.
- ↑ Frank S., Jankovic J. Advances in the Pharmacological Management of Huntington’s Disease (англ. ) // Drugs (англ.) (рус. : journal. — Adis International, 2010. — Vol. 70, no. 5. — P. 561—571. — doi:10.2165/11534430-000000000-00000. — PMID 20329804. Архивировано 8 октября 2011 года. Архивированная копия (неопр.) (недоступная ссылка). Дата обращения: 25 апреля 2011. Архивировано 8 октября 2011 года.
- ↑ Walker F. O. Huntington’s disease (англ.) // The Lancet. — Elsevier, 2007. — Vol. 369, no. 9557. — P. 224. — doi:10.1016/S0140-6736(07)60111-1. — PMID 17240289.
- ↑ FDA Approves First Drug for Treatment of Chorea in Huntington’s Disease (неопр.). FDA Approves First Drug for Treatment of Chorea in Huntington’s Disease. U.S. Food and Drug Administration (15 августа 2008). Дата обращения: 10 августа 2008. Архивировано 1 июня 2012 года.
- ↑ Walker F. O. Huntington’s disease (англ.) // The Lancet. — Elsevier, 2007. — Vol. 369, no. 9557. — P. 225. — doi:10.1016/S0140-6736(07)60111-1. — PMID 17240289.
- ↑ Huntington Disease (неопр.). genereviews bookshelf. University of Washington (19 июля 2007). Дата обращения: 12 марта 2009. Архивировано 10 февраля 2009 года.
- ↑ Bonelli R. M., Wenning G. K., Kapfhammer H. P. Huntington’s disease: present treatments and future therapeutic modalities (англ.) // International Clinical Psychopharmacology (англ.) (рус. : journal. — 2004. — Vol. 19, no. 2. — P. 51—62. — doi:10.1097/00004850-200403000-00001. — PMID 15076012.
- ↑ Обнаружено потенциальное направление для лечения болезни Гентингтона Архивная копия от 17 мая 2012 на Wayback Machine
- ↑ Лахинимод оказался неэффективен против болезни Хантингтона Архивная копия от 7 сентября 2018 на Wayback Machine remedium. ru, 06.09.2018
- ↑ Teva drops development of laquinimod Архивная копия от 7 сентября 2018 на Wayback Machine PharmaTimes Media, 06.09.2018
- ↑ Search of: Huntington Disease — List Results — ClinicalTrials.gov (англ.). ClinicalTrials.gov. Дата обращения: 11 ноября 2021. Архивировано 11 ноября 2021 года.
- ↑ Search: Huntington Disease — List Results — Active — ClinicalTrials.gov (англ.). Дата обращения: 11 ноября 2021. Архивировано 11 ноября 2021 года.
- ↑ Munoz-Sanjuan I, Bates GP (February 2011). “The importance of integrating basic and clinical research toward the development of new therapies for Huntington disease”. The Journal of Clinical Investigation [англ.]. 121 (2): 476—83. DOI:10.1172/JCI45364. PMC 3026740. PMID 21285520.
- ↑ McBride JL, Pitzer MR, Boudreau RL, Dufour B, Hobbs T, Ojeda SR, Davidson BL (December 2011). “Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington’s disease”. Molecular Therapy [англ.]. 19 (12): 2152—62. DOI:10.1038/mt.2011.219. PMC 3242667. PMID 22031240.
- ↑ Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, Pytel KA, Artates JW, Weiss A, Cheng SH, Shihabuddin LS, Hung G, Bennett CF, Cleveland DW (June 2012). “Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis”. Neuron [англ.]. 74 (6): 1031—44. DOI:10.1016/j.neuron.2012.05.009. PMC 3383626. PMID 22726834.
- ↑ Barnes DW, Whitley RJ (February 1987). “Antiviral therapy and pulmonary disease”. Chest [англ.]. 91 (2): 246—51. DOI:10.1172/JCI45130. PMC 3026739. PMID 3026739.
- ↑ Taube to fund $3m Huntington’s disease research in US, The Times of Israel (3 мая 2017). Архивировано 3 мая 2017 года. Дата обращения 23 ноября 2021.
Литература
- Эдвин Кёрк. Мальчик, который не переставал расти и другие истории про гены и людей = Edwin Kirk. The genes that makes us.. — М.: Альпина нон-фикшн, 2022. — 312 с. — ISBN 978-5-00139-405-1.
Болезнь Гентингтона – Симптомы и причины
Обзор
Болезнь Гентингтона – редкое наследственное заболевание, вызывающее прогрессирующее разрушение (дегенерацию) нервных клеток в головном мозге. Болезнь Гентингтона оказывает широкое влияние на функциональные возможности человека и обычно приводит к двигательным, мыслительным (когнитивным) и психическим расстройствам.
Симптомы болезни Гентингтона могут развиться в любое время, но часто они впервые появляются у людей в возрасте 30–40 лет. Если заболевание развивается до 20 лет, оно называется ювенильной болезнью Гентингтона. Когда болезнь Хантингтона развивается рано, симптомы несколько отличаются, и болезнь может прогрессировать быстрее.
Доступны лекарства, помогающие справиться с симптомами болезни Гентингтона. Но лечение не может предотвратить физическое, умственное и поведенческое ухудшение, связанное с этим заболеванием.
Товары и услуги
- Книга: Книга семейного здоровья клиники Мэйо, 5-е издание
- Информационный бюллетень: Письмо о здоровье клиники Мэйо — цифровое издание
Симптомы
Болезнь Хантингтона обычно вызывает двигательные, когнитивные и Признаки и симптомы. Какие симптомы появляются первыми, сильно различаются от человека к человеку. Некоторые симптомы кажутся более доминирующими или оказывают большее влияние на функциональные способности, но они могут меняться на протяжении болезни.
Двигательные расстройства
Двигательные расстройства, связанные с болезнью Гентингтона, могут включать как проблемы с непроизвольными движениями, так и нарушения произвольных движений, такие как:
- Непроизвольные подергивания или корчи (хорея)
- Проблемы с мышцами, такие как ригидность или контрактура мышц (дистония)
- Медленные или необычные движения глаз
- Нарушения походки, осанки и равновесия
- Трудности с речью или глотанием
Нарушения произвольных движений, а не непроизвольных движений, могут оказывать большее влияние на способность человека работать, выполнять повседневные действия, общаться и оставаться независимым.
Когнитивные расстройства
Когнитивные нарушения, часто связанные с болезнью Гентингтона, включают:
- Трудности с организацией, расстановкой приоритетов или сосредоточением внимания на задачах
- Отсутствие гибкости или склонность зацикливаться на мысли, поведении или действии (настойчивость)
- Отсутствие контроля над импульсами, что может привести к вспышкам, необдуманным действиям и сексуальной распущенности
- Отсутствие осознания собственного поведения и способностей
- Медленность в обработке мыслей или «поиске» слов
- Трудность в изучении новой информации
Психические расстройства
Наиболее распространенным психическим расстройством, связанным с болезнью Гентингтона, является депрессия. Это не просто реакция на получение диагноза болезни Гентингтона. Вместо этого депрессия, по-видимому, возникает из-за повреждения головного мозга и последующих изменений в его функции. Признаки и симптомы могут включать:
- Чувство раздражительности, грусти или апатии
- Социальный уход
- Бессонница
- Усталость и упадок сил
- Частые мысли о смерти, умирании или самоубийстве
Другие распространенные психические расстройства включают:
- Обсессивно-компульсивное расстройство, состояние, характеризующееся повторяющимися навязчивыми мыслями и повторяющимся поведением
- Мания, которая может вызывать повышенное настроение, гиперактивность, импульсивное поведение и завышенную самооценку
- Биполярное расстройство, состояние с чередующимися эпизодами депрессии и мании
В дополнение к вышеуказанным нарушениям у людей с болезнью Гентингтона часто наблюдается потеря веса, особенно по мере прогрессирования заболевания.
Симптомы юношеской болезни Гентингтона
Начало и прогрессирование болезни Гентингтона у молодых людей могут немного отличаться от таковых у взрослых. Проблемы, которые часто проявляются на ранних стадиях заболевания, включают:
Поведенческие изменения
- Трудности с концентрацией внимания
- Быстрое, значительное падение общей успеваемости в школе
- Поведенческие проблемы
Физические изменения
- Напряженные и ригидные мышцы, влияющие на походку (особенно у детей младшего возраста)
- Тремор или легкие непроизвольные движения
- Частые падения или неуклюжесть
- Изъятия
Когда обратиться к врачу
Обратитесь к врачу, если заметите изменения в движениях, эмоциональном состоянии или умственных способностях. Признаки и симптомы болезни Гентингтона могут быть вызваны рядом различных состояний. Поэтому важно получить своевременную и тщательную диагностику.
Записаться на прием в клинику Mayo
Из клиники Mayo на ваш почтовый ящик
Зарегистрируйтесь бесплатно и будьте в курсе последних научных достижений, советов по здоровью и актуальных тем, связанных со здоровьем, таких как COVID-19. плюс опыт управления здоровьем.
Чтобы предоставить вам самую актуальную и полезную информацию, а также понять, какая информация полезна, мы можем объединить вашу электронную почту и информацию об использовании веб-сайта с другая информация о вас, которой мы располагаем. Если вы пациент клиники Майо, это может включать защищенную информацию о здоровье. Если мы объединим эту информацию с вашей защищенной медицинской информации, мы будем рассматривать всю эту информацию как информацию и будет использовать или раскрывать эту информацию только так, как указано в нашем уведомлении о практики конфиденциальности. Вы можете отказаться от получения сообщений по электронной почте в любое время, нажав на ссылка для отписки в письме.
Причины
Аутосомно-доминантный тип наследования
Аутосомно-доминантный тип наследования
При аутосомно-доминантном заболевании измененный ген представляет собой доминантный ген, расположенный на одной из неполовых хромосом (аутосомах). Вам нужен только один измененный ген, чтобы быть затронутым этим типом расстройства. У человека с аутосомно-доминантным заболеванием — в данном случае у отца — есть 50-процентная вероятность рождения больного ребенка с одним измененным геном (доминантный ген) и 50-процентная вероятность рождения здорового ребенка с двумя типичными генами (рецессивные гены). ).
Болезнь Гентингтона вызывается наследственным отличием в одном гене. Болезнь Хантингтона является аутосомно-доминантным заболеванием, а это означает, что человеку для развития заболевания требуется только одна копия нетипичного гена.
За исключением генов половых хромосом, человек наследует две копии каждого гена — по одной копии от каждого родителя. Родитель с нетипичным геном может передать нетипичную копию гена или здоровую копию. Таким образом, каждый ребенок в семье имеет 50-процентную вероятность унаследовать ген, вызывающий генетическое заболевание.
Осложнения
После начала болезни Хантингтона функциональные возможности человека со временем постепенно ухудшаются. Скорость прогрессирования заболевания и продолжительность варьируют. Время от первых симптомов до смерти часто составляет от 10 до 30 лет. Болезнь ювенильного Гентингтона обычно приводит к смерти в течение 10 лет после появления симптомов.
Клиническая депрессия, связанная с болезнью Гентингтона, может увеличить риск самоубийства. Некоторые исследования показывают, что наибольший риск суицида возникает до постановки диагноза и на средних стадиях заболевания, когда человек начинает терять самостоятельность.
В конце концов, человеку с болезнью Гентингтона требуется помощь во всех повседневных делах и уход. На поздних стадиях болезни человек, скорее всего, будет прикован к постели и не сможет говорить. Люди с болезнью Гентингтона, как правило, способны понимать язык и хорошо осведомлены о семье и друзьях, хотя некоторые не узнают членов семьи.
Общие причины смерти включают:
- Пневмонию или другие инфекции
- Травмы, связанные с падениями
- Осложнения, связанные с неспособностью глотать
Профилактика
Экстракорпоральное оплодотворение
Экстракорпоральное оплодотворение
Во время экстракорпорального оплодотворения яйцеклетки удаляются из зрелых фолликулов в яичнике (А). Яйцеклетка оплодотворяется путем введения одного сперматозоида в яйцеклетку или смешивания яйцеклетки со спермой в чашке Петри (В). Оплодотворенная яйцеклетка (эмбрион) переносится в матку (С).
Люди с известным семейным анамнезом болезни Гентингтона по понятным причинам обеспокоены тем, что они могут передать ген Гентингтона своим детям. Эти люди могут рассмотреть варианты генетического тестирования и планирования семьи.
Если родитель из группы риска рассматривает генетическое тестирование, может быть полезно встретиться с консультантом по генетическим вопросам. Генетический консультант обсудит потенциальные риски положительного результата теста, который будет указывать на то, что у родителя разовьется заболевание. Кроме того, парам необходимо будет сделать дополнительный выбор в отношении того, иметь ли детей или рассмотреть альтернативы, такие как пренатальное тестирование на наличие гена или экстракорпоральное оплодотворение донорской спермой или яйцеклетками.
Другим вариантом для пар является экстракорпоральное оплодотворение и преимплантационная генетическая диагностика. В этом процессе яйцеклетки извлекаются из яичников и оплодотворяются спермой отца в лаборатории. Эмбрионы проверяются на наличие гена Хантингтона, и только те из них, у которых отрицательный результат на ген Хантингтона, имплантируют в матку матери.
от сотрудников клиники Майо
Связанные
Связанные процедуры
Продукты и услуги
О заболеваниях Хантингтона и связанных с ним расстройствах в больнице Джона Хопкинса
. Краткая история болезни ХантингтонаГЛАВО назван в честь Джорджа Хантингтона, который описал его среди жителей Ист-Хэмптона, Лонг-Айленд, в 1872 году. Это наследственное нейродегенеративное заболевание. В 1993 году совместная группа исследователей обнаружила ген, вызывающий БХ. В результате этого открытия теперь можно диагностировать БГ по образцам крови или тканей.
Генетическая причина БХ БХ вызывается мутацией в гене, расположенном на хромосоме 4. Этот ген находится у каждого человека и содержит последовательность CAG-повтора. Мы еще не открыли нормальную функцию гена. В случае БХ ген содержит аномально большое количество повторов CAG. Вообще говоря, чем больше количество триплетных повторов, тем раньше в жизни разовьется БХ. Кроме того, когда ген передается от отца к ребенку (но не от матери к ребенку), ген может удлиняться еще больше, что приводит к более раннему возрасту начала заболевания. Это явление известно как ожидание .
Гены болезней могут быть как доминантными, так и рецессивными. Ген HD является доминантным. У каждого ребенка больного родителя есть шанс 50/50 получить мутантный ген и, следовательно, 50% шанс унаследовать заболевание. С другой стороны, если люди, родители которых страдают БХ, не наследуют мутантный ген, они не могут передать его кому-либо еще.
Важная информация о HD-тестировании Важно понимать, что, хотя люди рождаются с мутировавшим геном БХ, в большинстве случаев у них симптомы не проявляются до более позднего возраста. Поэтому у кого-то может быть бессимптомное или предсимптомное течение нескольких лет. В прошлом не было возможности проверить наличие аномального гена, но теперь анализ крови может определить, является ли человек носителем гена БХ. Этот тест можно использовать в случаях подозрения на БХ для подтверждения диагноза или в качестве прогностического теста у лиц, подверженных риску БХ. Люди в группе риска могут захотеть пройти прогностическое тестирование, чтобы успокоиться, спланировать свои медицинские потребности или перед рождением детей. Решение о проведении такого теста является важным и не должно восприниматься легкомысленно. В большинстве центров, которые проводят прогностическое тестирование, в том числе и в нашем, требуется период консультирования до и после теста.
Начало обычно в среднем возрасте, но может возникнуть в любое время от детства до старости. Начальные признаки этого расстройства могут быть малозаметными. БГ характеризуется двигательным расстройством, деменцией и психическими расстройствами. Дополнительные характеристики БГ включают изменения личности, потерю веса (вероятно, из-за сочетания трудностей с приемом пищи и сжигания калорий при непроизвольных движениях), трудности с глотанием и трудную для понимания речь.
Как только у человека появляются признаки БГ, течение болезни может длиться от десяти до тридцати лет. Как правило, течение БГ можно условно разделить на три этапа.
Ранняя стадия:
На этой стадии пациенты все еще могут выполнять большинство своих обычных действий. Возможно, они все еще работают и могут водить машину. Непроизвольные движения мягкие и редкие, речь все еще четкая, а деменция, если она вообще присутствует, выражена слабо.
Средняя стадия:
На этой стадии пациенты более недееспособны и могут нуждаться в помощи в некоторых повседневных делах. Падения, потеря веса и трудности с глотанием могут стать проблемой. Слабоумие более очевидно для случайного наблюдателя. Непроизвольные движения более выражены.
Поздняя стадия:
Пациенты, вступившие в эту стадию заболевания, нуждаются почти в полном уходе и могут проживать в больницах или домах престарелых, хотя некоторые остаются дома. Они больше не могут ходить или говорить. Теперь они могут быть более жесткими и демонстрировать меньше непроизвольных движений. Люди на этой стадии могут или не могут глотать пищу. На этой стадии большинство пациентов, кажется, не сильно страдают, поскольку они, по-видимому, не осознают своего окружения. Когда пациент умирает, причина смерти обычно связана с недоеданием, пневмонией или сердечной недостаточностью.
В настоящее время лекарства от БГ нет. Исследователи работают над рядом методов лечения, которые могут замедлить прогрессирование заболевания. На сегодняшний день существует ряд вмешательств, которые улучшают качество жизни больных БГ. На ранних и средних стадиях заболевания больным ГБ можно давать небольшие дозы лекарств, чтобы помочь подавить непроизвольные движения. Депрессия и другие психические состояния у людей с ГБ поддаются достаточно эффективному лечению. Правильное питание, физические упражнения и меры предосторожности в домашних условиях могут помочь свести к минимуму многие потенциальные последствия БГ, такие как потеря веса, падения и удушье. Наконец, контакт с другими больными БГ, членами семьи и лицами, осуществляющими уход, может стать жизненно важным источником поддержки для пациентов с БГ и их семей.
Люди с HD должны обсуждать свои опасения и пожелания относительно лечения/вмешательств (например, зондов для кормления, запросов на реанимацию) и вскрытий со своими семьями и врачами, пока они еще могут говорить за себя.
Болезнь Хантингтона: MedlinePlus Genetics
Описание
Болезнь Гентингтона — это прогрессирующее заболевание головного мозга, вызывающее неконтролируемые движения, эмоциональные проблемы и потерю мыслительных способностей (познания).
Болезнь Хантингтона с началом во взрослом возрасте, наиболее распространенная форма этого расстройства, обычно появляется у человека в возрасте 30–40 лет. Ранние признаки и симптомы могут включать раздражительность, депрессию, мелкие непроизвольные движения, плохую координацию и проблемы с усвоением новой информации или принятием решений. У многих людей с болезнью Гентингтона развиваются непроизвольные подергивания или судорожные движения, известные как хорея. По мере прогрессирования заболевания эти движения становятся более выраженными. У пострадавших людей могут быть проблемы с ходьбой, речью и глотанием. Люди с этим расстройством также испытывают изменения в личности и снижение мыслительных и логических способностей. Люди с формой болезни Гентингтона, начинающейся у взрослых, обычно живут от 15 до 20 лет после появления первых признаков и симптомов.
Менее распространенная форма болезни Гентингтона, известная как ювенильная форма, начинается в детстве или подростковом возрасте. Это также связано с проблемами движения и психическими и эмоциональными изменениями. Дополнительные признаки ювенильной формы включают замедленность движений, неуклюжесть, частые падения, ригидность, невнятную речь и слюнотечение. Успеваемость в школе снижается по мере того, как ухудшаются мыслительные и логические способности. Судороги возникают у 30-50% детей с этим заболеванием. Ювенильная болезнь Гентингтона имеет тенденцию к более быстрому прогрессированию, чем взрослая форма; больные люди обычно живут от 10 до 15 лет после появления признаков и симптомов.
Частота
Болезнь Гентингтона поражает от 3 до 7 человек европейского происхождения на 100 000 человек. Расстройство, по-видимому, менее распространено в некоторых других группах населения, включая людей японского, китайского и африканского происхождения.
Причины
Мутации в гене HTT вызывают болезнь Гентингтона. Ген HTT предоставляет инструкции для создания белка, называемого хантингтином. Хотя функция этого белка неясна, похоже, что он играет важную роль в нервных клетках (нейронах) головного мозга.
Мутация HTT , вызывающая болезнь Гентингтона, включает сегмент ДНК, известный как тринуклеотидный повтор CAG. Этот сегмент состоит из серии трех строительных блоков ДНК (цитозин, аденин и гуанин), которые появляются несколько раз подряд. В норме сегмент CAG повторяется в пределах гена от 10 до 35 раз. У людей с болезнью Гентингтона сегмент CAG повторяется от 36 до более чем 120 раз. У людей с 36–39 CAG-повторами могут развиться или не развиться признаки и симптомы болезни Гентингтона, в то время как у людей с 40 или более повторами заболевание развивается почти всегда.
Увеличение размера CAG-сегмента приводит к продукции аномально длинной версии белка хантингтина. Удлиненный белок разрезается на более мелкие токсичные фрагменты, которые связываются друг с другом и накапливаются в нейронах, нарушая нормальные функции этих клеток. Дисфункция и возможная гибель нейронов в определенных областях мозга лежат в основе признаков и симптомов болезни Гентингтона.
Наследование
Это состояние наследуется по аутосомно-доминантному типу, что означает, что одной копии измененного гена в каждой клетке достаточно, чтобы вызвать заболевание. Больной человек обычно наследует измененный ген от одного больного родителя. В редких случаях у человека с болезнью Гентингтона нет родителя с этим заболеванием.
По мере того как измененный ген HTT передается из поколения в поколение, размер тринуклеотидного повтора CAG часто увеличивается. Большее количество повторов обычно связано с более ранним появлением признаков и симптомов. Это явление называется ожиданием. Люди со взрослой формой болезни Гентингтона обычно имеют от 40 до 50 повторов CAG в гене HTT , в то время как люди с ювенильной формой заболевания, как правило, имеют более 60 повторов CAG.
У лиц, имеющих от 27 до 35 повторов CAG в гене HTT , болезнь Гентингтона не развивается, но они подвержены риску рождения детей, у которых разовьется это заболевание. По мере передачи гена от родителя к ребенку размер тринуклеотидного повтора CAG может удлиняться до диапазона, связанного с болезнью Хантингтона (36 повторов или более).
Другие названия этого состояния
- Хорея Хантингтона
- Хроническая прогрессирующая наследственная хорея Хантингтона
- Хорея Гентингтона
- Болезнь Гентингтона
Дополнительная информация и ресурсы
Информация о генетическом тестировании
- Реестр генетического тестирования: болезнь Гентингтона
- Реестр генетического тестирования: Болезнь Гентингтона с началом в юношеском возрасте
Информационный центр генетических и редких заболеваний
- Болезнь Гентингтона
- Ювенильная болезнь Гентингтона
Ресурсы поддержки пациентов и защиты интересов
- Информационный поиск по болезням
- Национальная организация редких заболеваний (NORD)
Научные исследования от ClinicalTrials.
gov- ClinicalTrials.gov
Каталог генов и болезней от OMIM
- БОЛЕЗНЬ ГАНТИНГТОНА
Научные статьи в PubMed
- PubMed
Ссылки
- Бейтс GP. История генетических заболеваний: молекулярная генетика Хантингтона болезнь — история. Нат Рев Жене. 2005 г., 6 октября (10): 766-73. дои: 10.1038/nrg1686. Обзор. Цитата в PubMed
- Кэрон Н.С., Райт Г.Э.Б., Хейден М.Р. Болезнь Гентингтона. 1998 г., 23 октября [обновлено в 2020 г. 11 июня]. Пришли: Адам MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Грипп К.В., Амемия А., редакторы. GeneReviews® [Интернет]. Сиэтл (Вашингтон): университет Вашингтон, Сиэтл; 1993-2022 гг. Доступна с http://www.ncbi.nlm.nih.gov/books/NBK1305/ Цитата на PubMed
- Гонсалес-Алегре П., Афифи А.К. Клинические характеристики детского возраста
(юношеский) Болезнь Гентингтона: отчет о 12 пациентах и обзор
литература. J Чайлд Нейрол. 2006 март; 21(3):223-9. Цитата в PubMed
- Имарисио С., Кармайкл Дж., Корольчук В., Чен К.В., Сайки С., Роуз С., Кришна Г., Дэвис Дж. Э., Ттофи Э., Андервуд Б. Р., Рубинштейн Д. С. Болезнь Гентингтона: от патологии и генетики к потенциальным методам лечения. Biochem J. июнь 2008 г. 1;412(2):191-209. дои: 10.1042/BJ20071619. Обзор. Цитата в PubMed
- Джонс Л., Хьюз А. Патогенные механизмы болезни Гентингтона. Int Rev Нейробиол. 2011;98:373-418. doi: 10.1016/B978-0-12-381328-2.00015-8. Обзор. Цитата в PubMed
- Кент А. Болезнь Гентингтона. Стенд Нурс. 2004 21–27 апреля; 18 (32): 45–51; контрольный опрос 52-3. Обзор. Цитата в PubMed
- Maiuri T, Mocle AJ, Hung CL, Xia J, van Roon-Mom WM, Truant R. Хантингтин каркасный белок в комплексе ответа на окислительное повреждение ДНК ATM. Хум Мол Жене. 2017 15 января; 26(2):395-406. дои: 10.1093/hmg/ddw395. Цитата в PubMed
- Старрок А. , Ливитт Б.Р. Клинические и генетические особенности Гентингтона
болезнь. J Geriatr Psychiatry Neurol. 2010 дек.; 23(4):243-59. дои:
10.1177/0891988710383573. Epub 2010 5 октября. Обзор. Цитата в PubMed
- Тост Х., Вендт К.С., Шмитт А., Хайнц А., Браус Д.Ф. Болезнь Хантингтона: феноменологическое разнообразие нервно-психического состояния, которое бросает вызов традиционные концепции неврологии и психиатрии. Am J Психиатрия. 2004 г. Январь; 161 (1): 28-34. Цитата на PubMed
- Уокер ФО. Болезнь Хантингтона. Ланцет. 2007 20 января; 369 (9557): 218-28. Обзор. Цитата в PubMed
- Янг АБ. Хантингтин в норме и болезни. Джей Клин Инвест. 2003 г. Февраль; 111 (3): 299-302. Обзор. Цитирование в PubMed или бесплатная статья в PubMed Central
Жизнь с болезнью Гентингтона: «Для нашей семьи конец света всегда близок» | Семья
День, когда я узнала, что умру, начался достаточно безобидно: обычное размытие смены подгузников и раздражительных сообщений мужу. Жизнь в нашем недавно отреставрированном лондонском доме вошла в привычный ритм, с низким фоном домашнего недовольства. Споры об обоях исчерпали себя; наши кошки помирились с нашей годовалой дочерью Анной; и я была рада выйти замуж за ответственного гедониста, который любил детей, но никогда не заставлял меня чувствовать себя виноватой за то, что находил их скучными.
В тот день мой муж Том рано ушел на работу; режиссер-документалист, снимал сериал о лондонском метро. После бессонной ночи я завтракал с Анной, когда зазвонил стационарный телефон. Это был старый друг моего отца Эрик, который присматривал за ним с тех пор, как четыре года назад умерла моя мама. Мы все волновались, потому что Мерф (все называли моего папу Мерфом) принимал неверные решения, а затем вызывающе копался.
Эрик сказал, что мне следует кое-что знать: у Мерф диагностировали болезнь Гентингтона (БХ). Я никогда не слышал об этом заболевании и понятия не имел, что это такое. Так что, конечно же, я сделал то, чего никогда не следует делать: погуглил. В Википедии это звучит немного похоже на болезнь Альцгеймера и немного на болезнь Паркинсона, но хуже, чем и то, и другое. Это было дегенеративное неврологическое состояние, и звучало ужасно; бедный Мёрф. Хотя это имело смысл, учитывая некоторые вещи, которые я заметила в нем с тех пор, как умерла моя мать. Он был нервным и суетливым, и в его когда-то уверенной походке была какая-то нерешительность.
Потом немного о генетике. Говоря сухим языком Вики, «БГ наследуется по аутосомно-доминантному типу. Вероятность того, что каждый потомок унаследует пораженный ген, составляет 50%». Все, что случилось с моим отцом, могло случиться и со мной или с моим братом. И если бы у меня был этот ген, мои дети тоже были бы в опасности. В отличие от болезни Альцгеймера, БГ обычно проявляется в среднем возрасте, в возрасте от 35 до 45 лет. Мне было 35 лет.
Первым видимым признаком является хорея — судорожные, неконтролируемые, непроизвольные движения во всех частях тела. По мере дегенерации частей мозга пациенты страдают от серьезных когнитивных проблем: потери памяти, суждения, способности к самоорганизации. Им трудно ходить, и они склонны к падениям. Они теряют способность глотать и иногда умирают от недоедания. Часто страдает и их личность. Сообщения об агрессивном, компульсивном и неприемлемом сексуальном поведении являются обычным явлением. Ближе к концу семьи часто не видят другого выхода, кроме как поместить своих страдающих родственников в специализированные учреждения. Лекарства от болезни Гентингтона не существует.
Я позвонил Тому. Было около половины девятого, и он направлялся на съемочную площадку на станции Эджвар-Роуд. Я была в слезах, когда объясняла, что сказал мне Эрик и что я прочитала. Как обычно, он пытался меня успокоить, но на этот раз его настойчивость в том, что интернет просто обязан преувеличивать масштабы бедствия, была, к сожалению, неуместной.
Потом телефон отключился. Он прибыл на станцию через несколько минут после того, как в туннеле под поездом взорвалась бомба. Это было 7 июля 2005 года, и до конца дня он был вне связи, оказавшись в плену последствий самого страшного террористического акта, который когда-либо видела столица.
Я включил BBC News 24 и наблюдал за развитием событий. Я продолжал пробовать телефон Тома, но он переключался прямо на голосовую почту. Я не могу вспомнить многих подробностей того дня, но я знаю, как это было. Было ощущение, что наша жизнь потемнела по краям. Когда Том вернулся в зону досягаемости, я плакала от облегчения, но все еще не могла переварить ошеломившую нас новость.
Мой общий друг познакомил меня с Томом на вечеринке в Ноттинг-Хилле в 2002 году. интересно. Наша первая совместная ночь была пьяным смехом. В то время не было приложений для знакомств — мы просто занимались романтикой. Это было ударом и промахом, но более приятным для этого. Что бы Том написал в своем профиле? Нашел бы я его страсть к футбольному клубу «Фулхэм» отталкивающей? Был также его вкус в одежде: я был фашистом стиля с имиджем, который нужно поддерживать, и обычно меня не видели бы мертвым с кем-то в брюках Berghaus.
Мои потребности затмили все остальные. Мне нужно было, чтобы меня любили раньше детей, а это сложно, потому что я никогда не была более непривлекательной
Но когда он говорил, все остальные люди в комнате поблекли до серых тонов. Это действительно была любовь с первого разговора. Он даже заставил футбол казаться захватывающим. Мне было приятно видеть, как мои самоограничивающие убеждения исчезают между диванными подушками. У нас было так много общего. Его любимая песня была и моей любимой: Another Girl, Another Planet by the Only Ones. На тот момент небольшие области различий все еще казались интересными, а не угрожающими.
Мы целовались поверх пальто в комнате для гостей. Мы вернулись ко мне домой, и утром я захотел завтрак в Макдональдсе. У нас обоих было похмелье, но Том отправился на главную улицу в поисках яичных макмаффинов. Он заблудился на обратном пути. Я боялся, что он не вернется; было несколько тревожных минут отсутствия его. Эта маленькая сценка представила все основные темы наших последующих отношений. Тома воспитывали послушным; Я был воспитан властным и нуждающимся. Оглядываясь назад, я хотел бы одеться, а не лежать, как царица Савская. Мы могли бы пойти в Макдональдс вместе. То, что могло бы начаться как совместное партнерство, с самого начала было перекошено в мою пользу.
Через несколько дней я встретил друга и сказал ему, что встретил человека, за которого собираюсь выйти замуж. Раньше я никогда не думал о том, чтобы жениться на ком-либо. Просматривая все свои отношения, я мог видеть, что ни один из моих бывших не стал бы хорошим отцом, даже если бы они были женатыми. Когда я решила это, я подумала, что это будет лишь вопросом времени, когда Том будет убежден силой аргументов, что я подходящая ему женщина.
Год спустя мы с Томом были в Скай на мини-отпуске с подругой и ее будущим бывшим мужем. Все выходные мы спорили. Мне не терпелось романтических признаний или полного объяснения, почему он еще не сделал предложение. Неужели он теперь знал обо мне все?
У меня была форма, когда нужно было требовать подарки, прежде чем их можно было дарить бесплатно. Мерф всегда давал мне деньги на мой день рождения, который приходится на сентябрь. Вместо того, чтобы ждать, я попросил бы его об этом в августе или июле. В Скае я хотел, чтобы Том продвигал свое предложение руки и сердца точно так же. Но мое нытье возымело обратный эффект (никто не любит, когда его режут), и мы чуть не расстались. По дороге домой все выглядело рискованно, но затем, в зоне выдачи багажа, вероятно, все еще испытывая ко мне двойственное чувство, он достал бриллиант, который был у него все это время при себе. Когда мы сели в машину, мы оба почувствовали себя обманутыми из-за того момента, который он запланировал как воспоминание, которым мы будем дорожить. Но это была моя вина.
Теперь, спустя 15 лет после того, как мне поставили диагноз, я задаюсь вопросом, было ли такое неразумное и, в конечном счете, пагубное поведение продуктом моего воспитания, болезнью Гентингтона или их комбинацией. Гораздо легче обвинить HD, чем себя, но я не могу отделаться от мысли, что у меня не было никаких симптомов, когда я ужасно себя вел с Томом. Возможно, я преждевременно потерял эмпатию из-за болезни, но это не оправдание для того, чтобы бросать свой вес.
Мы поженились в 2003 году в отеле Old Ship в Брайтоне. С того момента, как я заказал организаторов свадьбы, и до последней песни вечеринки, сценарий представлял собой фантазию, ориентированную на невесту, в которой Том был маргинализирован. Мы согласились, что лучше всего подойдет Брайтон, и обоим понравился Старый Корабль. Но это было последнее решение, которое мы приняли вместе.
Мое несогласие с его общественной жизнью обсуждалось в каждом конкретном случае. Кто-то, с кем он познакомился в университете и стал интернет-предпринимателем, был изгнан по политическим мотивам. Другая подруга хотела, чтобы мы играли в салонные игры на ее 30-летие, поэтому я заставила Тома отвезти меня домой (я ненавижу салонные игры). Его семья играла в Consequences на Рождество, что я презирал (единственные рождественские традиции в моей семье — слишком много пить и смотреть телевизор). Так что я бы отсидел, тыкая.
Я не хотел, чтобы он развлекался, если он не был со мной или в моем окружении. Когда ему удавалось отвлечься на вечер, я навязчиво писала ему сообщения. Было душно и угнетающе. Если бы мы пошли гулять с его друзьями, я бы оделся так, чтобы убить всех в зоне досягаемости.
У нас было много места, но я хотел увеличить его. Я нашел свою идею постоянного дома на прекрасном угловом участке на улице с цветущей вишней на деревьях. Пока мы делали ремонт, я узнала, что беременна Анной.
Кто-то однажды описал HD как болезнь скорби, что кажется очень уместным. Вы теряете свою индивидуальность и часть своей человечности, оставаясь при этом достаточно осознанным, чтобы подсчитывать каждую потерю. После того, как я узнал о Мерфе, я решил пройти тест, чтобы узнать, есть ли у меня этот ген. Я предполагал, что большинство на моем месте сделают то же самое, поэтому я был удивлен, узнав, что 95% из них этого не делают. Для меня желание пройти тест было рациональным ответом, а уклонение от него — трусливой отговоркой.
Когда я узнал, что у меня есть ген HD, что мне оставалось делать, как не плакать? Даже стойкий Том выглядел пораженным.
Для меня все началось с мелких необъяснимых пропаж: ключи от машины, очки, миллион зажигалок, обувь, одежда. Потом я потерял мир, город за городом. Знакомые места превратились в страшный клубок улиц, поэтому я остался в доме. Потом стала пропадать и сама машина: когда мы останавливались на сервисах, я никак не могла найти к ней дорогу. Затем последовали большие человеческие потери. Я потерял свою сексуальность. Друзья перестали вспоминать меня в гости. А потом я начал терять собственное прошлое: по мере того, как моя кратковременная и долговременная память подвергалась воздействию БГ, история моей жизни отступала вдаль и становилась все более недоступной для меня.
До всех этих потерь я жил настолько полноценно, насколько это возможно, с перспективой полного краха личности. Наш сын Джон родился в прекрасный день в апреле 2009 года, через три года после того, как мой тест дал положительный результат, и до появления симптомов. Нам потребовалось много времени, чтобы решить, заводить ли нам еще одного ребенка, зная, что у них будет 50% шанс унаследовать генную мутацию. Но в конечном счете я не хотел, чтобы Анна была единственным ребенком, справлялась со странным мной и не с кем было играть.
Мой терапевт продолжала плакать, когда я рассказывал ей о жизни с HD, но я не плакал. Прощение и принятие себя казались неуловимыми
Люди с болезнью Гентингтона иногда могут казаться безразличными и легкомысленными. Их явное пренебрежение эмоциональными потребностями партнера может быть болезненным, особенно когда это контрастирует с прежними любящими отношениями; естественная склонность партнера чувствовать себя обиженным. В этих ситуациях человек с БГ не ведет себя преднамеренно неуклюже, своевольно или недоброжелательно — его кажущаяся эгоцентричность является следствием потери психической гибкости, связанной с болезнью. Возможно, они больше не смогут поставить себя на место другого или взвесить обе стороны в споре. Они могут искренне не видеть, как их действия влияют на других.
Из-за моих различных недугов наше хозяйство не ладилось, пока мы не нашли сиделку, Эйд. Она также помогла с администрированием и моим списком дел. Но эмпатию нельзя передать на аутсорсинг; моя семья нуждалась в матери, которая могла бы общаться с ними, и я часто был слишком поглощен собой. Том воспитывал нас двоих (а также зарабатывал все деньги). Он вспомнил их набор для физкультуры, вовлек их в творческие занятия, такие как выпечка, чтобы они не сидели перед телевизором весь день, контролировал их текстовые сообщения и водил Анну на футбол. Что еще более важно, он был рядом с ними эмоционально, когда я был наполовину отстранен. Мне нельзя было доверять их недельный график; Я даже забыл рождественское шоу Джона. («Ты была единственной мумией, которой там не было. Единственной мумией!»)
Если я чувствую себя виноватым, значит, у меня есть надежда. Мог бы я так долго размышлять о своей бесчеловечности, если бы я действительно был бесчеловечен? Я читал рассказы людей, страдающих БГ, которые не стыдятся того, что ведут себя неподобающим образом или тратят деньги на автомобили, которые они не могут себе позволить, в то время как меня постоянно мучает чувство вины за опрометчивые покупки и то, что я дерьмовый родитель. Я приветствую это, как признак жизни.
«Он ежедневно составлял списки дел, чтобы никто из нас ничего не забыл». Фотография: Томас Даффилд/The Guardian У Тома есть все основания полагать, что я преждевременно потерял эмпатию. Наши отношения не были ни любовью, ни сотрудничеством с самого начала, поэтому было мало резервов доброй воли, на которые можно было бы опереться, когда HD позвонила.
Перенесемся на пару лет вперед, и я доедал молоко до того, как другие успели добраться до него, и отправлял Тома в магазин за добавкой — каждый день. Я никогда не спал и следил за тем, чтобы никто не спал. Мои потребности затмевали все остальные, и когда я хотел чего-то, я должен был это иметь сейчас . Меня нужно было кормить, любить и слушать перед детьми, что было непростой задачей, поскольку я никогда не была более непривлекательной.
Отсутствие сочувствия нанесло смертельный удар моему браку. Накануне одной из рабочих поездок Том всегда был суетливым, поэтому не было ничего удивительного, когда однажды утром в 2014 году он вывез весь мусор с нашей садовой свалки на свалку в наемном фургоне. Он починил канализацию, позвонил газовщикам по поводу протечки, водил Джона на урок гимнастики, косил газон, готовил на неделю, развешивал все белье и составлял ежедневные списки дел, чтобы никто из нас не мог забыть. ничего, пока он был в отъезде. Через несколько дней мне пришло в голову, что это могла быть генеральная репетиция его отъезда навсегда, и так оно и оказалось.
Мы договорились жить отдельно. Том заслужил второй шанс быть близким с кем-то, кто все еще способен на близость. Однако дело было не только в этом: HD не только лишил меня сексуальности, но и притупил мои эмоции. Том пообещал, что позаботится о том, чтобы у меня было достаточно поддержки, и он был верен своему слову, но иметь рядом людей, которые помогают с практическими делами и составляют мне компанию по вечерам, — это не то же самое, что иметь мужа.
Мы составили план, как сообщить детям, что разлучаемся, но в итоге я проигнорировал его — я всегда старался быть с ними откровенным и чувствовал, что должен учитывать тот факт, что я был мрачнее, чем обычный. Анна едва оторвалась от телефона: «Это было довольно очевидно, мама». Она сказала, что хочет жить со мной, и, похоже, была рада, что мы с Томом ладим, в отличие от разведенных родителей ее лучшей подруги, которые не могли находиться в одной комнате друг с другом.
Джон очень чувствительный, поэтому я был опечален, но не удивлен, когда он сказал мне, как его задели наши ссоры: «Ты думал, что я был наверху, когда ты кричал на папу, но я был за дверью». Затем, однажды, когда мы возвращались из школы, он сказал: «Если вы с папой разведетесь, это будет означать, что леди Хантингтона будет предоставлена сама себе». Мы с Анной заверили его, что Том всегда будет заботиться обо мне, даже если мы больше не будем жить вместе, но, конечно, он все равно беспокоился.
Что касается меня, то я был убит горем и местами возмущен. Как, черт возьми, он мог оставить меня? Однако в глубине души я понимал, что если бы я лучше относился к нему до того, как заболел, мы бы до сих пор были вместе. Возможно, он оставался со мной так долго только потому, что был воспитан в послушании до уровня самоуничижения — если бы не HD, он, вероятно, оставил бы меня много лет назад.
Том переехал в квартиру за углом; как ни странно, мы никогда не ладили лучше. Он сказал, что отпустить его было актом любви, который будет вдохновлять Анну и Джона на долгие годы. Но всегда было ужасно, когда он возвращался домой, в свою квартиру. Я не могла вынести мысли о том, что он может быть с кем-то еще, не в последнюю очередь потому, что они должны были быть необычайно умными и чуткими, чтобы понять нашу нетрадиционную установку — солидную женщину, которая чувствовала себя соперницей, когда я смотрел на нее. Потом я начал к этому привыкать. Остается только принять новую норму.
Субботний журнал
ШоуЭта статья взята из нового печатного журнала The Guardian, посвященного субботе, в котором лучшие материалы, культура, стиль жизни и статьи о путешествиях сочетаются в одной красивой упаковке. Доступно сейчас в Великобритании и ROI.
Фото: GNM
Было ли это полезно?
Хотел бы я воспитать его, когда у меня была возможность, но уже слишком поздно для сожалений. Я не хочу чувствовать себя отягощенным чувством вины за то, что я забыл о его дне рождения, и вижу только один способ избежать этого: помнить об этом с этого момента. Я хочу отпраздновать то хорошее, что у нас еще есть.
Джон сочувствует мне, поднимая вещи, когда я их роняю (хотя, к сожалению, ему все равно нужно платить за выполнение любой домашней работы, которая занимает больше трех минут). Анна, Том и даже семья Тома сочувствуют мне, постоянно уверяя, что я ни в чем не виноват. Но почему-то сейчас я не могу сочувствовать себе. Мой терапевт продолжала плакать, когда я рассказал ей о конце моего брака и ежедневных разочарованиях жизни с HD, но я этого не сделал. Я видел, что правильно работать над преодолением своей вины, но прощение и принятие себя казались недостижимыми.
Когда все улеглось, я начал думать, что проделал не такую уж плохую работу, учитывая ту руку, которую мне выдали. Я помогала заботиться о своей семье и держала ее на плаву в трудных обстоятельствах, не стонала и не стремилась снова оказаться на вершине счета. Я нашла это неожиданно полезным и не воспринимала материнство как понижение в должности, как могла бы когда-то. Я начала писать книгу в кабинете рядом с кухней и обнаружила, что меня совершенно не беспокоит шум детей, входящих и выходящих со своими друзьями.
Мой HD пошел дальше, и мои отношения изменились. После четырех лет разлуки вручили документы о разводе. Будь я в лучшем настроении, можно было бы устроить праздник. В мой день рождения Том сказал, какой прекрасной матерью я была, по его мнению, и что Анна превратилась в интеллектуально строгого и чуткого подростка, которым мы все гордимся. Джон полюбил исполнительское искусство и ходит в танцевальную школу, каждый день самостоятельно пользуясь общественным транспортом. По дороге домой он пишет мне, спрашивая, как прошел мой день, и бывает много моментов, подобных этому, когда я чувствую его присутствие. Иногда я задаюсь вопросом, связано ли его беспокойство с тем фактом, что он осознал мою смертность — и свою собственную — в таком юном возрасте. Для нашей семьи конец света всегда близок. Анна хочет провериться на этот ген, когда станет достаточно взрослой, но я заверил их обоих, что к тому времени будут доступны лекарства, которые лечат болезнь, а не только ее симптомы. Детская любовь окутала меня позитивом, без которого я бы не смогла обойтись. Между тем книга подошла к концу, и этот писательский проект изменил мое состояние. Я был наблюдателем за жизнью других людей дольше, чем я надеялся, наблюдая, как вокруг меня происходят вечеринки. Теперь, наконец, я автор своей судьбы.
Выдержки из книги «Пациент 1: забывая и обретая себя» Шарлотты Рэйвен, опубликованной Джонатаном Кейпом 4 ноября по цене 14,99 фунтов стерлингов. Чтобы поддержать Guardian и Observer, закажите свой экземпляр на guardianbookshop.com. Может взиматься плата за доставку.
За информацией и консультацией обращайтесь в Ассоциацию болезней Хантингтона. др. Каков исход ревмокардита у детей с хореей Сиденгама? Турок J Педиатр . 2012 март-апрель. 54(2):159-67. [Ссылка QxMD MEDLINE].
Ford JB, Albertson TE, Owen KP, Sutter ME, McKinney WB. Острая устойчивая хорея у детей после сверхтерапевтического приема препаратов, производных амфетамина. Педиатр Нейрол . 2012 сен. 47 (3): 216-8. [Ссылка QxMD MEDLINE].
Torreggiani S, Torcoletti M, Cuoco F, Di Landro G, Petaccia A, Corona F. Хорея, малоизвестное проявление системной красной волчанки: краткий обзор литературы и четыре клинических случая. Pediatr Rheumatol Online J . 2013 16 окт. 11(1):36. [Ссылка QxMD MEDLINE]. [Полный текст].
Твардовский А.О., Пас Х.А., Пасторино А.С., Джейкоб К.М., Маркес-Диас М.Дж., Сильва К.А. Хорея у ребенка с синдромом Чарга-Стросса. Acta Reumatol Port . 2010 январь-март. 35(1):72-5. [Ссылка QxMD MEDLINE].
Чидл ВБ. Различные проявления ревматического состояния при обследовании в детстве и молодости. Ланцет . 1889. 1:821-827, 871-877.
Ридель К.Р., Липпс Т.Д., Гилберт Д.Л. Распространенность нервно-психических расстройств при хорее Сиденгама. Педиатр Нейрол . 2010 апр. 42 (4): 243-8. [Ссылка QxMD MEDLINE].
Арон А.М., Фримен Дж.М., Картер С. Естественная история хореи Сиденхама. Обзор литературы и долгосрочная оценка с акцентом на сердечные последствия. Am J Med . 1965 янв. 38:83-95. [Ссылка QxMD MEDLINE].
Специальная писательская группа AHA. Руководство по диагностике ревматизма. Критерии Джонса, 1992 обновление. Специальная писательская группа Комитета по ревматической лихорадке, эндокардиту и болезни Кавасаки Совета по сердечно-сосудистым заболеваниям у молодежи Американской кардиологической ассоциации. ЯМА . 1992, 21 октября. 268(15):2069-73. [Ссылка QxMD MEDLINE].
Пунуколлу М., Мушет Н., Линни М., Хеннесси С., Мортон М. Нейропсихиатрические проявления хореи Сиденхама: систематический обзор. Dev Med Детский нейрол . 2016 янв. 58 (1):16-28. [Ссылка QxMD MEDLINE].
ван Иммерзел ТД, ван Гилст Р.М., Хартвиг Н.Г. Благотворное использование иммуноглобулинов при лечении хореи Сиденгама. Евро J Педиатр . 2010 сен. 169(9):1151-4. [Ссылка QxMD MEDLINE]. [Полный текст].
Giedd JN, Rapoport JL, Kruesi MJ, et al. Хорея Сиденгама: магнитно-резонансная томография базальных ганглиев. Неврология . 1995 г., декабрь 45 (12): 2199-202. [Ссылка QxMD MEDLINE].
Уокер К., Бринк А., Лоуренсон Дж., Матиассен В., Уилмшерст Дж.М. Лечение хореи Сиденгама внутривенным иммуноглобулином. J Детский нейрол . 2012 г. 27 февраля (2): 147-55. [Ссылка QxMD MEDLINE].
Гарви Массачусетс, Сведо SE. Хорея Сиденгама. Клинические и терапевтические обновления. Adv Exp Med Biol . 1997. 418:115-20. [Ссылка QxMD MEDLINE].
Letort D, Gonzalez-Alegre P. Болезнь Гентингтона у детей. Handb Clin Neurol . 2013. 113:1913-7. [Ссылка QxMD MEDLINE].
Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. Ген болезни Вильсона представляет собой предполагаемую транспортирующую медь АТФазу Р-типа, аналогичную гену Менкеса. Нат Жене . 1993, декабрь 5(4):327-37. [Ссылка QxMD MEDLINE].
Демиркиран М., Янкович Дж. Пароксизмальные дискинезии: клиника и классификация. Энн Нейрол . 1995 г., 38 октября (4): 571–579. [Ссылка QxMD MEDLINE].
Goodenough DJ, Fariello RG, Annis BL и др. Семейные и приобретенные пароксизмальные дискинезии. Предлагаемая классификация с описанием клинических признаков. Арка Нейрол . 1978 г., декабрь 35 (12): 827-31. [Ссылка QxMD MEDLINE].
Новый ген, содержащий тринуклеотидный повтор, который разрастается и нестабилен на хромосомах, вызывающих болезнь Хантингтона. Совместная исследовательская группа по болезни Гентингтона. Сотовый . 1993 26 марта. 72(6):971-83. [Ссылка QxMD MEDLINE].
Aasly J, Skandsen T, Rø M. Нейроакантоцитоз — вариабельность симптомов у двух братьев и сестер. Акта Нейрол Сканд . 1999 ноябрь 100(5):322-5. [Ссылка QxMD MEDLINE].
Аббас С., Ханна С., Тали А.Б. Обсессивно-компульсивное расстройство и ревматическая хорея: есть ли связь? Психопатология . 1996. 29(3):193-7. [Ссылка QxMD MEDLINE].
Бескинд Д.Л., Кейм С.М. Хореоатетозное двигательное расстройство у мальчика с Mycoplasma pneumoniae энцефалитом. Энн Эмерг Мед . 1994 г. 23 июня (6): 1375-8. [Ссылка QxMD MEDLINE].
Bird MT, Palkes H, Prensky AL. Последующее исследование хореи Сиденхама. Неврология . 1976 г. 26 июля (7): 601-6. [Ссылка QxMD MEDLINE].
Bruyn GW, Myrianthopoulos NC. Хроническая ювенильная наследственная хорея. Винкен П.Х., Брюн Г.В., Клаванс Х.Л., ред. Справочник по клинической неврологии . Амстердам: Издательство Elsevier Science; 1986. Том 49: 335-8.
Кэмпбелл М., Грега Д.М., Грин В.Х. и др. Нейролептические дискинезии у детей. Клин Нейрофармакол . 1983. 6(3):207-22. [Ссылка QxMD MEDLINE].
Сервера Р., Ашерсон Р.А., Фонт Дж. и др. Хорея при антифосфолипидном синдроме. Клинические, рентгенологические и иммунологические характеристики 50 пациентов из наших клиник и новейшей литературы. Медицина (Балтимор) . 1997 май. 76(3):203-12. [Ссылка QxMD MEDLINE].
Чедвик Д., Рейнольдс Э.Х., Марсден К.Д. Антиконвульсивные дискинезии: сравнение с дискинезиями, вызванными нейролептиками. J Нейрол Нейрохирург Психиатрия . 1976 г., декабрь 39 (12): 1210-8. [Ссылка QxMD MEDLINE].
Детский уход за здоровьем Dev. Дискинезии и связанные с ними психические расстройства после стрептококковых инфекций. Детский уход за здоровьем Dev . 11 2004. 30(6):729.
Comunale JP Jr, Heier LA, Chutorian AM. Ювенильная форма болезни Гентингтона: картина МРТ. AJR Am J Рентгенол . 1995 авг. 165(2):414-5. [Ссылка QxMD MEDLINE].
Дауд А.С., Заки М., Шакир Р. и др. Эффективность вальпроата натрия при лечении хореи Сиденгама. Неврология . 1990 г., июль 40 (7): 1140-1. [Ссылка QxMD MEDLINE].
де Кром MC, Борей AM, Харди EL. [Отравление марганцем из-за употребления таблеток Chien Pu Wan]. Нед Тайдшр Генескд . 1994 1 октября. 138(40):2010-2. [Ссылка QxMD MEDLINE].
Дрейк М.Э. младший, Джексон Р.Д., Миллер, Калифорния. Пароксизмальный хореоатетоз после черепно-мозговой травмы. J Нейрол Нейрохирург Психиатрия . 1986 г., июль 49(7):837-8. [Ссылка QxMD MEDLINE].
Фан С. Пароксизмальные дискинезии. Компакт-диск Марсдена, Фан С., ред. Двигательные расстройства 3 . Оксфорд, Англия: Баттерворт-Хайнеманн; 1994. 75-6.
Фаррер Л.А., Коннелли П.М. Генетическая модель возраста начала болезни Гентингтона. Am J Hum Genet . 1985 г. 37 марта (2): 350-7. [Ссылка QxMD MEDLINE].
Folstein SE, Chase GA, Wahl WE, et al. Болезнь Хантингтона в Мэриленде: клинические аспекты расовой изменчивости. Am J Hum Genet . 1987 авг. 41(2):168-79. [Ссылка QxMD MEDLINE].
Гаскон Г.Г., аль-Джараллах А.А., Окамото Э. и др. Хорея как проявление рецидива герпетического энцефалита. Мозговой разработчик . 1993 май-июнь. 15(3):178-81. [Ссылка QxMD MEDLINE].
Гордис Л. Фактическое исчезновение ревматизма в Соединенных Штатах: уроки подъема и падения болезни. Лекция памяти Т. Дакетта Джонса. Тираж . 1985 г., декабрь 72(6):1155-62. [Ссылка QxMD MEDLINE].
Gualtieri CT, Quade D, Hicks RE, et al. Поздняя дискинезия и другие клинические последствия лечения нейролептиками у детей и подростков. Am J Психиатрия . 1984 г., янв. 141(1):20-3. [Ссылка QxMD MEDLINE].
Хаддерс-Алгра М., Бос А.Ф., Мартейн А. и др. Детская хорея у младенца с тяжелой бронхолегочной дисплазией: исследование ЭМГ. Dev Med Детский нейрол . 1994 г. 36 февраля (2): 177-82. [Ссылка QxMD MEDLINE].
Hall JG, Te-Juatco L. Связь между возрастом начала и родительским наследованием при хорее Гентингтона. Am J Med Genet . 1983 16 октября (2): 289-90. [Ссылка QxMD MEDLINE].
Харди Р.Дж., Пуллон Х.В., Хардинг А.Е. и др. Нейроакантоцитоз. Клиническое, гематологическое и патологическое исследование 19случаи. Мозг . 1991, февраль 114 (часть 1A): 13–49. [Ссылка QxMD MEDLINE].
Hefter H, Mayer P, Benecke R. Стойкая хорея после рецидивирующей гипогликемии. Отчет о случае. Евро Нейрол . 1993. 33(3):244-7. [Ссылка QxMD MEDLINE].
Homan RW, Vasko MR, Blaw M. Концентрации фенитоина в плазме при пароксизмальном кинезигенном хореоатетозе. Неврология . 1980 30 июня (6): 673-6. [Ссылка QxMD MEDLINE].
Хантингтон Г. О хорее. Медицинский хирургический представитель . 1872. 26:317-321.
Husby G, van de Rijn I, Zabriskie JB, et al. Антитела, реагирующие с цитоплазмой нейронов субталамического и хвостатого ядер при хорее и острой ревматической лихорадке. J Exp Med . 1976 г., 1 октября. 144(4):1094-110. [Ссылка QxMD MEDLINE].
IHA и WFN. Руководство по молекулярно-генетическому прогностическому тесту при болезни Гентингтона. Исследовательская группа Международной ассоциации Хантингтона (IHA) и Всемирной федерации неврологов (WFN) по хорее Гентингтона. Неврология . 1994 авг. 44 (8): 1533-6. [Ссылка QxMD MEDLINE].
Иллариошкин С.Н., Игараши С., Онодера О. и др. Длина тринуклеотидных повторов и скорость прогрессирования болезни Гентингтона. Энн Нейрол . 1994 36 октября (4): 630-5. [Ссылка QxMD MEDLINE].
Икбал С., Шер М.Р., Гуд Р.А. и др. Разнообразие проявлений системной красной волчанки у детей. J Педиатр . 1999 окт. 135(4):500-5. [Ссылка QxMD MEDLINE].
Жубер Дж., Жубер П.Х. Хорея и психические изменения при отравлении фосфорорганическими соединениями. Отчет о еще 2 случаях. S Afr Med J . 1988 г., 2 июля. 74 (1): 32–4. [Ссылка QxMD MEDLINE].
Кашихара К., Ябуки С. [Односторонние хореические движения при идиопатическом гипопаратиреозе]. Нет Синкею . 1992 май. 44(5):477-80. [Ссылка QxMD MEDLINE].
Казис А., Кимискидис В., Георгиадис Г., Володаки Е. Нейроакантоцитоз с эпилепсией. Дж Нейрол . 1995 июнь 242(6):415-7. [Ссылка QxMD MEDLINE].
Кертес А. Пароксизмальный кинезигенный хореоатетоз. Сущность в рамках синдрома пароксизмального хореоатетоза. Описание 10 дел, в том числе 1 вскрытия. Неврология . 1967 г., 17 июля (7): 680–90. [Ссылка QxMD MEDLINE].
Kieburtz K, MacDonald M, Shih C, et al. Длина тринуклеотидных повторов и прогрессирование заболевания при болезни Гентингтона. Дж Мед Жене . 1994 31 ноября (11): 872-4. [Ссылка QxMD MEDLINE].
Конагая М., Конагая Ю. МРТ гемибализма вследствие хореи Сиденгама. J Нейрол Нейрохирург Психиатрия . 1992 март 55 (3): 238-9. [Ссылка QxMD MEDLINE].
Корн-Любецки I, Бранд А. Хорея Сиденхама в Иерусалиме: все еще присутствует. ISR Med Assoc J . 2004 г. 6 августа (8): 460-2. [Ссылка QxMD MEDLINE].
Korn-Lubetzki I, Brand A, Steiner I. Рецидив хореи Сиденгама: последствия для патогенеза. Арка Нейрол . 2004 авг. 61 (8): 1261-4. [Ссылка QxMD MEDLINE].
Krauss JK, Mohadjer M, Nobbe F, et al. Двусторонний баллизм у детей. Детская нервная система . 1991 7 октября (6): 342-6. [Ссылка QxMD MEDLINE].
Лели Д.А., Ферлоу Т.В. младший, Фальгоут Дж.К. Доброкачественная семейная хорея: связь с нарушением интеллекта. J Нейрол Нейрохирург Психиатрия . 1984 май. 47(5):471-4. [Ссылка QxMD MEDLINE].
Льюис-Джонсон Дж. Хорея: ее номенклатура, этиология и эпидемиология в клиническом материале округа Мальмхус. Acta Pediatr . 1949. 76:1910-1944.
MacMillan JC, Morrison PJ, Nevin NC, et al. Идентификация расширенного CAG-повтора в гене болезни Гентингтона (IT15) в семье, у которой сообщалось о доброкачественной наследственной хорее. Ж Мед Жене . 1993 г., 30 декабря (12): 1012-3. [Ссылка QxMD MEDLINE].
MacMillan JC, Snell RG, Tyler A, et al. Молекулярный анализ и клинические корреляции мутации болезни Гентингтона. Ланцет . 1993, 16 октября. 342(8877):954-8. [Ссылка QxMD MEDLINE].
Марк МХ. Двигательные расстройства: неврологические принципы и практика . Макгроу-Хилл; 1997. 38: 526-539.
Маркес-Диас М.Дж., Меркаданте М.Т., Такер Д. и др. Хорея Сиденгама. Психиатрическая клиника North Am . 1997 Декабрь 20 (4): 809-20. [Ссылка QxMD MEDLINE].
Mayeux R, Fahn S. Пароксизмальный дистонический хореоатетоз у пациента с семейной атаксией. Неврология . 1982 32 октября (10): 1184-6. [Ссылка QxMD MEDLINE].
Москет Б., Стараче Дж., Мадлен С. и др. Хорея-атетозный синдром под действием карбамазепина и вилоксазина. Последствия лекарственного взаимодействия?]. Терапия . 1994 ноябрь-декабрь. 49(6): 513-4. [Ссылка QxMD MEDLINE].
Myers RH, Goldman D, Bird ED, et al. Материнская передача при болезни Гентингтона. Ланцет . 1983 г., 29 января. 1(8318):208-10. [Ссылка QxMD MEDLINE].
Myers RH, Madden JJ, Teague JL, Falek A. Факторы, связанные с возрастом начала болезни Гентингтона. Am J Hum Genet . 1982 май. 34(3):481-8. [Ссылка QxMD MEDLINE].
Наусиеда П.А., Беляускас Л.А., Бэкон Л.Д. и др. Хроническая дофаминергическая чувствительность после хореи Сиденгама. Неврология . 1983 июнь 33(6):750-4. [Ссылка QxMD MEDLINE].
Наусиеда П.А., Гроссман Б.Дж., Коллер В.К. и др. Хорея Сиденгама: обновление. Неврология . 1980 30 марта (3): 331-4. [Ссылка QxMD MEDLINE].
Nomoto M, Thompson PD, Sheehy MP, et al. Антихолинергическая хорея в лечении фокальной дистонии. Мов Беспорядок . 1987. 2(1):53-6. [Ссылка QxMD MEDLINE].
Полсон Г. В., Рейдер К.Р. Двигательные расстройства у детей. В: Уоттс Р.Л., Коллер В.К., ред. Двигательные расстройства: неврологические принципы и практика. Том 49. Макгроу-Хилл. 1997: 61-672.
Perez-Duenas B, Prior C, Ma Q, Fernandez-Alvarez E, Setoain X, Artuch R. Детская хорея с церебральной гипотрофией: поддающийся лечению синдром энергетической недостаточности GLUT1. Арка Нейрол . 2009 ноябрь 66(11):1410-4. [Ссылка QxMD MEDLINE].
Пикколо И., Каусарано Р., Стерзи Р. и др. Хорея у больных СПИДом. Акта Нейрол Сканд . 1999 ноябрь 100(5):332-6. [Ссылка QxMD MEDLINE].
Pincus JH, Chutorian A. Семейная доброкачественная хорея с интенционным тремором: клиническая форма. J Педиатр . 1967 май. 70(5):724-9. [Ссылка QxMD MEDLINE].
Pozzan GB, Battistella PA, Rigon F, et al. Гипертиреоидная хорея у девочки-подростка. Мозговая разработка . 1992 март 14 (2): 126-7. [Ссылка QxMD MEDLINE].
Profumo R, Toce S, Kotagal S. Хореоатетоз новорожденных после пренатального воздействия оральных контрацептивов. Педиатрия . 1990, октябрь 86 (4): 648-9. [Ссылка QxMD MEDLINE].
Ринне Дж.О., Даниэль С.Е., Скаравилли Ф. и др. Нейропатологические особенности нейроакантоцитоза. Мов Беспорядок . 1994 май. 9(3):297-304. [Ссылка QxMD MEDLINE].
Родопман-Арман А., Язган Ю., Беркем М. и др. Присутствуют ли сенсорные феномены при хорее Сиденгама? Оценка 13 дел. Нейропиатрия . 2004 г. 35 августа (4): 242-5. [Ссылка QxMD MEDLINE].
Роос Р.А., Винцен А.Р., Виельвое Г. и др. Пароксизмальный кинезигенный хореоатетоз как симптом рассеянного склероза. J Нейрол Нейрохирург Психиатрия . 1991 г., июль 54(7):657-8. [Ссылка QxMD MEDLINE].
Rubio JP, Danek A, Stone C, et al. Хорея-акантоцитоз: генетическая связь с хромосомой 9q21. Am J Hum Genet . 1997 г., октябрь 61 (4): 899-908. [Ссылка QxMD MEDLINE].
Шейнберг И.Х., Штернлиб И. Болезнь Вильсона . Филадельфия: В. Б. Сондерс; 1984.
Шеннон К.М., Фенихель Г.М. Лечение хореи Сиденгама пимозидом. Неврология . 1990 янв. 40 (1): 186. [Ссылка QxMD MEDLINE].
Шульман Л.М., Сингер С., Вайнер В.Дж. Фенитоин-индуцированная фокальная хорея. Мов Беспорядок . 1996 11 января (1): 111-4. [Ссылка QxMD MEDLINE].
Stone LA, Armstrong RM. Необычное проявление диабета: гипергликемия, вызывающая гемибаллизм (абстр.). Энн Нейрол . 1989. 26:146.
Суховерский О. , Хейден М.Р., Мартин В.Р. и др. Мозговой метаболизм глюкозы при доброкачественной наследственной хорее. Мов Беспорядок . 1986. 1(1):33-44. [Ссылка QxMD MEDLINE].
Swedo SE, Leonard HL, Schapiro MB и др. Хорея Сиденхама: физические и психологические симптомы танца Святого Вита. Педиатрия . 1993 г., апрель 91(4):706-13. [Ссылка QxMD MEDLINE].
Swedo SE, Rapoport JL, Cheslow DL, et al. Высокая распространенность обсессивно-компульсивных симптомов у больных хореей Сиденгама. Am J Психиатрия . 1989 фев. 146(2):246-9. [Ссылка QxMD MEDLINE].
Sydenham T. Schedula monitoria de novae febris ingressu. Лондини, Г. Кеттилби. 1686.
Хорея Тибо Ф. Сиденхама. Винкен П.Дж., Брюн Г.В., ред. Справочник по клинической неврологии . Амстердам: Издательство Северной Голландии; 1968. 409-434.
Tison FX, Ferrer X, Julien J. Отсроченные двигательные расстройства как осложнение центрального миелинолиза моста. Мов Беспорядок . 1991. 6(2):171-3. [Ссылка QxMD MEDLINE].
Truong DD, Harding AE, Scaravilli F, et al. Двигательные нарушения при митохондриальных миопатиях. Исследование девяти случаев с двумя вскрытиями. Мов Беспорядок . 1990. 5(2):109-17. [Ссылка QxMD MEDLINE].
Vance JM, Pericak-Vance MA, Bowman MH, et al. Хорея-акантоцитоз: отчет о трех новых семьях и последствиях для генетического консультирования. Am J Med Genet . 1987 28 октября (2): 403-10. [Ссылка QxMD MEDLINE].
Veasy LG, Wiedmeier SE, Orsmond GS и др. Рецидив острой ревматической лихорадки в межгорной зоне США. N Английский J Med . 1987 г., 19 февраля. 316(8):421-7. [Ссылка QxMD MEDLINE].
Verheul GA, Tyssen CC. Рассеянный склероз, протекающий с пароксизмальной односторонней дистонией. Мов Беспорядок . 1990. 5(4):352-3. [Ссылка QxMD MEDLINE].
Болезнь Уолша М. Вильсона (HLD). Винкен П.Дж., Брюн Г.В., ред. Справочник по клинической неврологии . Амстердам: Издательство Северной Голландии; 1976. Том 27: 379-414.
Болезнь Уолша М. Вильсона: клинические симптомы. Арч Ди Чайлд . 1962. 37:253-6.
Ван Б.Дж., Чанг Ю.К. Терапевтические уровни фенитоина в крови при лечении пароксизмального хореоатетоза. Мониторинг лекарств . 1985. 7(1):81-2. [Ссылка QxMD MEDLINE].
Went L. Заявление о политике по вопросам этики в отношении молекулярно-генетического прогностического теста на болезнь Гентингтона. Международная ассоциация Хантингтона. Всемирная федерация неврологов. Дж Мед Жене . 1990 27 января (1): 34-8. [Ссылка QxMD MEDLINE].
Уилер П.Г., Уивер Д.Д., Добинс В.Б. Доброкачественная наследственная хорея. Педиатр Нейрол . 1993 сентябрь-октябрь. 9(5):337-40. [Ссылка QxMD MEDLINE].
Yimaz Y, Kocaman C, Ozdemir N. Карбамазепин в лечении хореи. Медицинский журнал Мармара . 2006. 19(1):27-29.
Zacchetti O, Sozzi G, Zampollo A. Пароксизмальный кинезигенный хореоатетоз. История болезни. Ital J Neurol Sci . 1983 Сентябрь 4 (3): 345-7. [Ссылка QxMD MEDLINE].
Хорея: признаки, причины и лечение
Хорея — двигательное расстройство, вызывающее непроизвольные, непредсказуемые движения тела.
Симптомы хореи могут варьироваться от незначительных движений, таких как ерзание, до серьезных неконтролируемых движений рук и ног. Также может мешать:
- речь
- глотание
- осанка
- походка
Симптомы хореи обычно зависят от состояния, вызвавшего ее. Обычный симптом — «хватка доярки». Люди с этим заболеванием не имеют скоординированных мышц рук и сжимают и отпускают руку, как будто доят. Еще один симптом – непроизвольное высовывание языка.
Хорея Движения могут быть быстрыми или медленными. Может показаться, что человек корчится от боли и не имеет телесного контроля. Эти движения также называют танцевальными или похожими на игру на фортепиано.
Состояния, связанные с хореей, и ее симптомы включают:
Болезнь Хантингтона
Болезнь Гентингтона является наследственным заболеванием. Это вызывает разрушение нервных клеток в вашем мозгу. Люди с болезнью Гентингтона могут испытывать симптомы хореи, такие как непроизвольные подергивания или корчи. Хватка доярки также является распространенным симптомом.
Хорея чаще встречается у людей с болезнью Гентингтона во взрослом возрасте. Со временем симптомы могут ухудшиться, а движения могут повлиять на ноги и руки.
Хорея-акантоцитоз
Это очень редкое генетическое заболевание. Для него характерны деформированные эритроциты. Это вызывает неврологические аномалии и влияет на работу мозга.
Хорея при этом состоянии обычно включает:
- ненормальные движения рук и ног
- пожимание плечами
- толчки тазом
Также может включать быстрые, бесцельные движения лица.
У людей с этой формой хореи также может наблюдаться дистония. Это характеризуется непроизвольными сокращениями мышц рта и лица, такими как:
- Зубной шлифование
- Необеспеченная отрыжка
- Слюни или плевание
- Укусы губы и языка
- Сложность с речью или общением
- Сложность Шальновые
- . Помимо хореи и дистонии, это состояние может вызвать:
- судороги
- невропатию
- потерю чувствительности
- мышечную слабость
- изменения поведения и личности
Хорея Сиденгама
Хорея Сиденгама в основном поражает детей и подростков. Это следует за стрептококковой инфекцией. Это также может быть осложнением ревматизма.
Этот тип хореи в основном поражает:
- лицо
- руки
- кисти
Он может препятствовать произвольным движениям, затрудняя выполнение основных задач, таких как одевание или кормление.
Это также может привести к:
- частому падению предметов
- ненормальной походке
- мышечной слабости
- невнятной речи
- снижению мышечного тонуса
Еще один распространенный симптом называется «язык арлекина». Когда человек с этим симптомом пытается высунуть язык, вместо этого язык выскакивает и высовывается.
Люди с ревматизмом в анамнезе чаще страдают хореей. Другие факторы риска связаны с риском развития конкретного заболевания.
Например, болезнь Гентингтона — это наследственное заболевание, которое может вызывать хорею. По данным клиники Майо, у человека, у родителей которого есть болезнь Гентингтона, 50-процентный шанс унаследовать болезнь.
Хорея связана с несколькими дополнительными причинами, как временными, так и хроническими. Эти причины включают:
- СПИД
- генетические состояния, такие как болезнь Гентингтона
- иммунные состояния, такие как системная красная волчанка
- инфекционные состояния, такие как хорея Сиденгама
- лекарственные препараты, включая леводопу и нейролептики
- метаболические или эндокринные нарушения, включая гипогликемию
- беременность, известная как хорея беременных тщательный сбор анамнеза для определения возможных причин. Чтобы диагностировать хорею, врач может спросить:
- Когда появились симптомы?
- Что улучшает или ухудшает симптомы? Ваши симптомы хореи имеют тенденцию ухудшаться, когда вы испытываете стресс?
- Есть ли у вас семейная история болезни Гентингтона?
- Какие лекарства вы принимаете?
Некоторые лабораторные анализы могут указывать на наличие хореи. Например, аномальные уровни меди в организме могут указывать на болезнь Вильсона — генетическое заболевание, вызывающее хорею.
Анализы на остроконечные эритроциты или эритроциты могут свидетельствовать о хорее-акантоцитозе. Анализы крови на гормоны паращитовидной железы или гормоны щитовидной железы могут указывать на метаболическую или эндокринную хорею.
При болезни Гентингтона визуализирующие исследования, такие как МРТ, могут показать активность мозга, которая является индикатором заболевания.
Лечение хореи зависит от ее типа. Он направлен на лечение основного заболевания, которое поможет при симптомах хореи.
Например, хорея Сиденгама поддается лечению антибиотиками. Хорея при болезни Гентингтона может лечиться нейролептиками, а также другими лекарствами.
Хорея, вызванная болезнью Паркинсона, неизлечима, но ее симптомы можно контролировать.
Лекарства
Большинство лекарств от хореи влияют на дофамин. Дофамин — это нейротрансмиттер или химическое вещество мозга, которое, среди прочего, контролирует движение, мышление и удовольствие в вашем мозгу.
Многие двигательные расстройства связаны с уровнем дофамина. Эти расстройства включают болезнь Паркинсона и синдром беспокойных ног.
Некоторые лекарства блокируют дофаминовые рецепторы, поэтому ваше тело не может использовать это химическое вещество. Многие из них являются антипсихотическими препаратами, которые, по-видимому, уменьшают хорею. К этим препаратам, которые врачи могут назначать не по прямому назначению, относятся:
- Fluphenazine (Prolixin)
- Haloperidol (Haldol)
- оланзапин (Zyprexa)
- кветиапин (серокекл)
- Риспер -(Risperdal)
99.
Диспериновый (Risperdal)
9. (Ксеназин). Лекарства, известные как бензодиазепины, такие как клоназепам (клонопин), также могут помочь уменьшить хорею.
Противосудорожные препараты, уменьшающие спонтанные движения, также могут уменьшать симптомы хореи.
Операции
Глубокая стимуляция головного мозга — это хирургический подход, который обещает лечение хореи. Это лечение включает в себя имплантацию электродов в мозг для регулирования нервных импульсов.
Если хорея не поддается лечению, врач может порекомендовать глубокую стимуляцию мозга. Эта процедура не излечивает хорею, но может уменьшить ее симптомы.
Уход на дому
Хорея повышает вероятность падения человека. Меры по уходу на дому включают установку нескользких поверхностей на лестницах и в ванных комнатах для предотвращения травм. Поговорите со своим врачом о других способах изменить свой дом в целях безопасности.
Перспективы хореи зависят от состояния, вызвавшего ее.