
Воспалительная миопатия – Воспалительная миопатия — причины, симптомы, диагностика и лечение
Воспалительная миопатия — причины, симптомы, диагностика и лечение
Воспалительная миопатия — воспалительный процесс, возникающий преимущественно в скелетных мышцах и приводящий к их дегенеративным изменениям. Воспалительная миопатия характеризуется мышечной слабостью, болями в мышцах, резким снижением объема активных движений, развитием контрактур, уплотнением и отечностью мышц. Из методов диагностики воспалительной миопатии наиболее информативно определение уровня КФК и миоглобина, электромиография и биопсия мышц. Лечение воспалительной миопатии осуществляется высокими дозами глюкокортикостероидов с последующей поддерживающей терапией. Однако достаточно часто встречаются резистентные к кортикостероидам формы заболевания.
Общие сведения
Воспалительные миопатии представляют собой целую группу заболеваний, в которую входят как системные поражение мышечной ткани, так и локальные воспалительные процессы в отдельных мышцах. Воспалительная миопатия системного характера представлена дерматомиозитом, полимиозитом, эозинофильным миозитом, миозитом с включениями, ревматической полимиалгией и миопатиями при системных заболеваниях. Примером локальной воспалительной миопатии может быть миозит глазных мышц, псевдотромбофлебит мышц голени и др. Кроме того, воспалительная миопатия может наблюдаться при некоторых инфекционных заболеваниях.
Возраст пациентов, наиболее подверженных заболеваемости, различается в зависимости от вида воспалительной миопатии. Так, полимиозит обычно возникает у лиц старше 30 лет. Дерматомиозит наблюдается как у взрослых, так и в детском возрасте. А миозит с включениями чаще встречается после 40 лет и является наиболее распространенной формой воспалительной миопатии в пожилом возрасте.
Воспалительная миопатия
Причины возникновения воспалительной миопатии
В некоторых случаях воспалительная миопатия напрямую связана с инфекционным процессом. Агентами, вызывающими инфекционно-воспалительно поражение мышц, могут быть вирусы (краснуха, грипп, ВИЧ-инфекция, энтеровирусы и пр.), бактерии (чаще стрептококковые инфекции), паразитарные инвазии (цистицеркоз, токсоплазмоз, трихинеллез). При этом заболевание классифицируется как инфекционная воспалительная миопатия.
В тех случаях, когда прямая связь миопатии с инфекцией не прослеживается, говорят об идиопатической миопатии. К ней относятся: миозит с включениями, полимиозит, дерматомиозит, миопатии при системных заболеваниях (системной красной волчанке, склеродермии, синдроме Шегрена, системных васкулитах, ревматоидном артрите). Наиболее распространенным как в неврологии, так и в ревматологии является мнение, что идиопатическая воспалительная миопатия имеет аутоиммунный механизм развития. Однако до сих пор не выделен антиген, который запускает лежащую в ее основе аутоиммунную реакцию. В отдельных случаях воспалительная миопатия имеет семейный характер, свидетельствующий о ее генетическом происхождении.
Симптомы воспалительной миопатии
Воспалительная миопатия клинически проявляется болями в мышцах, прогрессирующей мышечной слабостью и тугоподвижностью в пораженных отделах конечностей. Болевой синдром (миалгия) возникает не только в период двигательной активности, но и при прощупывании мышц, а иногда и в состоянии полного покоя. Мышечная слабость, как правило, начинает проявляться затруднением при удерживании предметов в руках. По мере ее прогрессирования для пациента становится невозможным поднять руку или ногу, сесть, встать. Воспалительная миопатия распространенного характера часто приводит к значительному сокращению объема активных движений, которые может выполнять больной, а иногда и к полной обездвиженности.
Воспалительная миопатия протекает с отеком и уплотнением пораженных мышц. В результате воспаления в них происходят атрофические изменения, развивается миофиброз — замещение мышечных волокон на соединительнотканные, возможен кальциноз. Образующиеся мышечные контрактуры приводят к тугоподвижности суставов. Зачастую воспалительная миопатия имеет длительное ремиттирующее течение. В некоторых случаях, чаще у детей, наблюдается полное выздоровление после перенесенного острого приступа воспалительной миопатии.
Наиболее тяжело воспалительная миопатия протекает при вовлечении в воспалительный процесс мышц гортани и дыхательной мускулатуры, что ведет к развитию миопатического пареза гортани и дыхательной недостаточности. Слабость дыхательных мышц приводит к снижению легочной вентиляции и возникновению застойной пневмонии. Слабость мышц глотки является причиной дисфагии (расстройств глотания), при которой возможно попадание пищи в дыхательные пути и возникновение аспирационной пневмонии. Если воспалительная миопатия распространяется на глазодвигательные мышцы, то возникает косоглазие и опущение верхнего века. Поражение мимических мышц приводит к маскообразному выражению лица. Системная воспалительная миопатия, например, дерматомиозит и полимиозит, может сопровождаться поражением сердечной мышцы с клиникой миокардита, кардиомиопатии и сердечной недостаточности.
Полимиозит проявляется типичным для воспалительной миопатии мышечным синдромом, который захватывает мышцы проксимальных отделов конечностей, в первую очередь плечевого и тазового пояса. В большинстве случаев наблюдается также поражение внутренних органов: ЖКТ (диарея, запор, кишечная непроходимость, язва желудка с перфорацией или желудочно-кишечным кровотечением), сердца и сосудов (артериальная гипотония, аритмия, кардиомиопатия и др), легких (очаговая или долевая пневмония). В 15% случаев эта воспалительная миопатия сопровождается суставным синдромом в виде артритов суставов кисти, реже — других суставов конечностей.
Дерматомиозит имеет сходную с полимиозитом клиническую картину. Отличительной чертой является наличие кожных проявлений: эритематозных пятен, участков гипо- и гиперпигментации, очагов гиперкератоза. Возможно поражение слизистых оболочек с развитием стоматита, гингивита, конъюнктивита.
Миозит с включениями характеризуется ранним поражением мускулатуры дистальных отделов рук: сгибателей пальцев и мышц предплечья. В ногах воспалительная миопатия распространяется как на дистальные, так и на проксимальные мышечные группы. Типично поражение разгибателей стопы и четырехглавой мышцы.
Диагностика воспалительной миопатии
К диагностике воспалительной миопатии помимо неврологов могут привлекаться ревматологи, кардиологи, пульмонологи, дерматологи, иммунологи и др. специалисты. К сожалению, на сегодняшний день воспалительная миопатия не имеет четких диагностических критериев. Определенное значение имеет обнаружение в биохимическом анализе крови повышения уровня креатинфосфокиназы (КФК), а также лактатдегидрогеназы и альдолазы, что свидетельствует о повреждении мышечной ткани. Зачастую воспалительная миопатия сопровождается повышением миоглобина. Однако уровень этих показателей не коррелирует с тяжестью клинических проявлений. По этой причине единственным достоверным критерием прогрессирования миопатии могут быть лишь результате исследования мышечной силы (эргометрии) в динамике. Следует также отметить, что повышение КФК и миоглобина наблюдается далеко не при всех видах воспалительной миопатии. Например, при миозите с включениями уровень КФК и миоглобина часто находится в пределах нормы.
Важное диагностическое значение имеют данные электромиографии. При дерматомиозите и полимиозите наблюдается снижение продолжительности и амплитуды потенциалов двигательных единиц, повышенная электровозбудимость. При миозите с включениями наряду с типичными для миопатии низкоамплитудными и кратковременными потенциалами отмечаются высокоамплитудные и длительные потенциалы, характеризующие нейрогенное поражение.
Уточнить характер поражения мышц и определить его распространенность позволяет биопсия мышц. Морфологическое исследование биоптата выявляет наличие воспалительных инфильтратов, атрофические изменения и некроз мышечных волокон, их замещение соединительной тканью.
Диагностика сопутствующих поражений внутренних органов осуществляется при помощи рентгенографии легких, ЭКГ, Эхо-КГ, гастроскопии, УЗИ органов брюшной полости.
Лечение воспалительной миопатии
Общепринятым методом лечения воспалительной миопатии является терапия глюкокортикостероидами. Суточная доза преднизолона составляет 80-100 мг. При достижении клинического эффекта лечения в виде увеличения мышечной силы производят постепенное снижение суточной дозы до поддерживающего уровня — 15 мг/сут. В тяжелых случаях воспалительная миопатия лечится пульс-терапией метилпреднизолоном. Сложности применения длительного лечения глюкокортикоидами состоит в их побочных эффектах, из-за которых подобная терапия противопоказана пациентам с язвенной болезнью и язвой желудка, артериальной гипертензией, остеопорозом, катарактой и глаукомой. Отсутствие увеличения мышечной силы в течение 3 месяцев с момента начала лечения кортикостероидами говорит о стероидной резистентности воспалительной миопатии.
Альтернативными препаратами в лечении воспалительной миопатии являются цитостатики (азатиоприн, метотрексат, циклоспорин, циклофосфамид). Они применяются вместо кортикостероидов при наличии стероидной резистентности или в комбинации с ними для уменьшения дозы глюкокортикостероидов во избежание их побочных эффектов.
Поскольку системная воспалительная миопатия обычно имеет аутоиммунный характер, то в ее лечении зачастую применяется плазмаферез, позволяющий снизить количество ЦИК в крови.
www.krasotaimedicina.ru
причины, симптомы, диагностика и лечение
Воспалительные миопатии — гетерогенная группа редких приобретенных заболеваний мышц. Характеризуется наличием воспалительного процесса в скелетных (поперечнополосатых) мышцах, вследствие которого происходят дегенеративные изменения. При воспалительных миопатиях воспаление носит аутоиммунный или инфекционный характер. Данный вид миопатии сопровождается:
- мышечной слабостью;
- болями в мышцах;
- атрофиями;
- наличием контрактур;
- понижением двигательной активности;
- возникновением отечности мышц;
- в некоторых случаях наблюдается мышечное уплотнение.
Содержание статьи:
Воспалительные миопатии — это большая группа заболеваний, включающая в себя системные поражения мышечной ткани (дерматомиозит, полимиозит, эозинофильный миозит, миозит с включениями, ревматическая полимиалгия), а также локальные воспаления в отдельных группах мышц (миозит глазных мышц, псевдотромбофлебит мышц голени и пр.).
Возраст людей, когда наиболее высок риск инфицирования воспалительными миопатиями, зависит от типа заболевания. Например, полимиозит наблюдается чаще после тридцати лет, миозит с включениями поражает людей старше сорока лет, являясь при этом самой распространённой формой воспалительных миопатий, встречающихся в пожилом возрасте.
Причины возникновения воспалительной миопатии
Причин возникновения воспалительных миопатий достаточно много. Врачи акцентируют внимание на том, что этология и патогенез заболеваний представленной группы до конца не изучены, не имеют четкой схемы.
- Некоторые инфекционные заболевания (грипп, ВИЧ-инфекции, краснуха и пр.) могут стать провокаторами воспалительной миопатии.
- Причинами, вызывающими воспалительные миопатии, могут стать также стрептококковые бактерии, паразитарные инвазии вроде токсоплазмоза или трихинеллёза, а также грибковые инфекции.
- Существуют также идиопатические миопатии (заболевания, возбуждение которых не связано с инфекциями), они имеют аутоиммунный механизм развития, но следует сказать, что антиген, который лежит в основе возбуждения аутоиммунной реакции, так и не вычислен. При изучении заболевания выявлены семейные случаи, исходя из чего можно судитьо генетической природе данной группы заболеваний.
К идиопатическим воспалительным миопатиям относят: полимиозит, миозит с включениями, миопатии при системных заболеваниях (системной красной волчанке, склеродермии, синдроме Шегрена, ревматоидном артрите, системных воскулитах). Пациенты с дерматомиозитом и полимиозитом чаще подвержены онкологическим заболеваниям, особенно в виде злокачественных опухолей.
Симптомы воспалительной миопатии
Воспалительные миопатии имеют следующую симптоматику:
- Болевой синдром в мышцах (миалгия). При прогрессировании болезни боли наблюдаются не только во время двигательной активности, но и в период покоя или при незначительном надавливании на мышцы.
- Мышечная слабость. Человек испытывает трудности при сжатии предметов в руках. Прогрессируя, заболевание делает почти невозможным поднятие руки или ноги, лишает больного возможности самостоятельно садиться и вставать.
- Значительное сокращение объема активных движений, а в отдельных случаях пациент приковывается к кровати.
- Отек и уплотнение пораженных мышц.
- Развитие атрофии и миофиброза (мышечные волокна замещаются на соединительнотканные).
- Кальциноз.
- Контрактуры.
- Тугоподвижность суставов.
- Миопатический парез гортани.
- Дыхательная недостаточность.
- Застойная и аспирационная пневмония (возникают как следствие слабости дыхательных мышц и снижения легочной вентиляции).
Клиническая картина заболеваний из группы воспалительных миопатий различна, как и продолжительность протекания. При дерматомиозите, например, симптомы начинают проявляться в виде общего недомогания и повышенной температуры, лихорадки. Впоследствии наблюдается характерная сыпь на кожных покровах, а затем слабость проксимальных мышц. Сыпь фиолетового цвета может наблюдаться на поверхностях век, сопровождается отечностью и телангиоэктазиями на шее и груди, между фалангами пальцев и на кисти — эритематозная сыпь. На суставах коленей и локтей появляется утолщение кожи. При дерматомиозите наблюдается гиперемия щек, дисколорация и утолщение ногтевых лож, что приводит к отечности пальцев. Далее, прогрессируя, болезнь приводит к тугоподвижности мышц и болевому синдрому. Происходит поражение в основном проксимальных мышц, дистальные мышцы менее поддаются травматизации.
У детей заболевание может проявлять себя как острый приступ или чередование обострений и ремиссий. При быстром прогрессировании может возникнуть дыхательная недостаточность, поскольку происходит поражение мышц гортани и дыхательных путей. У большей половины инфицированных детей были выявлены кальцификаты в подкожных тканях. В отдельных случаях болезнь заканчивается летально (около 10% всех инфицированных детей).
Почти у половины взрослых пациентов выявляются злокачественные опухоли при диагностике дерматомиозита (для сравнения у больных полимиозитом опухоли наблюдаются очень редко). В результате, симптоматика онкологического образования накладывается на клиническую картину дерматомиозита, что существенно усложняет диагностику и лечение заболевания. При удалении злокачественной опухоли происходит регресс мышечной слабости. Истинная причина злокачественных новообразований в клинической картине дерматомиозита остается неизвестной.
Диагностика воспалительной миопатии
Диагностика воспалительных миопатий затруднена. Это связано с широким разбросом клинической картины. Симптомы очень схожи с заболеваниями, которые относятся к группе воспалительных миопатий, а также заболеваниями иной природы. В процессе диагностики необходимо, в первую очередь, дифференцировать заболевание среди однородных ему. Пациент должен пройти консультации у невропатолога, дерматолога, пульмонолога и кардиолога для получения наиболее точной клинической картины и исключения похожих заболеваний.
При большинстве воспалительных миопатий (кроме миозита с включениями) может наблюдаться повышение СОЭ в крови, но почти у половины этот показатель показывает норму. Важным показателем при диагностике данного заболевания будет КФК (уровень креатинфосфокиназы), по которому можно судить о степени повреждения мышц и мышечной мембраны. Зачастую можно наблюдать повышения уровня изофермента КФК. Концентрация миоглобина в сыворотке тоже повышается и свидетельствует о явном прогрессировании заболевания и назначении адекватного лечения.
Диагностируя данное заболевания, назначают также ЭМГ, результаты которого важны для установления диагноза. При полимиозите и дерматомиозите, обычно наблюдаются кратковременные полифазные потенциалы двигательных единиц (это касается проксимальных мышц). Похожие изменения можно наблюдать также при миозите с включениями. Кроме того, похожие аномалии могут быть выявлены в процессе ЭМГ и при нейрогенном поражении. Поэтому установить точный диагноз, полагаясь лишь на ЭМГ, невозможно. Проводят также биопсию мышц. Этот анализ помогает определить распространённость воспалительного процесса.
Проводится гормональное исследование при отсутствии сыпи. Целью данного исследования будет определение содержания трийодтиронина, тиреотропного гормона TSH и тироксина.
Лечение воспалительной миопатии
Патогенического и этиологического лечения не существует. Лечение в основном направлено на устранение симптомов и общее поддержание мышечной силы, устранение контрактур, а также на достижение медикаментозной ремиссии.
Несмотря на ограниченное количество информации о способе лечения данных заболеваний, врачи пришли к единому мнению — иммуносупрессивная терапия дает хорошие результаты. Чаще всего пациенту назначается целый комплекс мероприятий. Во-первых, это медикаментозное терапевтическое лечение. Одной из самый успешных и эффективных методик является глюкокортикоидная терапия (преднизолон и метипред). Доза препаратов — 1 мг на 1 кг веса. Принимают препараты строго через день во избежание осложнений, которые могут появиться вследствие применения глюкокортикоидной терапии. Назначая такую терапию, врач должен тщательно обследовать пациента на наличие сопутствующих заболеваний, которые могут вызвать осложнения. К таким заболеваниям, при которых глюкокортикоидная терапия противопоказана относятся:
- сахарный диабет;
- остеопороз;
- язва желудка;
- ожирение или любые другие нарушения обмена веществ;
- повышенное артериальное давление.
Ремиссия наступает приблизительно через один-два месяца. После этого доза препаратов уменьшается под четким контролем игольчатой ЭМГ. Иногда лечащим врачом принимается решение о замене базового препарата при условии значительных улучшений в анализе клинической картины. Чаще всего препаратом-заменителем выступает азатиоприн. Если глюкокортикоидная терапия не дала желаемых результатов, то для усиления эффекта параллельно назначается цитостатическая терапия.
Для скорейшей ремиссии врачи советуют также лечебную физкультуру (строго под присмотром методиста по ЛФК или компетентного специалиста), проведение курса массажей, различные ортопедические мероприятия. Врачи рекомендуют активное движение, длительное лежание противопоказано. Преждевременное использование кресла-каталки также не рекомендуется, поскольку предусматривает покой значительной части мышц.
Говоря о прогнозах данной группы заболеваний, то они неплохие. При своевременном и адекватном лечении около половины больных существенно улучшают свое общее состояние и полностью излечиваются. Треть пациентов остается с ощущением мышечной слабости. В течение первых трех-четырех лет примерно 15% пациентов с тяжёлой формой умирают от сопутствующих заболеваний сердца, легких и пр. Значительная часть смертельных случаев происходит по причине образовавшихся онкологических опухолей.
www.mosmedportal.ru
Воспалительные миопатии: причины, симптомы, диагностика, лечение
Воспалительные миопатии представляют собой группу заболеваний, которые вызваны с хроническим мышечным воспалением, слабостью мышц и, в отдельных случаях, мышечными спазмами и болью. Миопатия — общее медицинское понятие, используемое для описания дистрофии мышц. Все миопатии приводят к мышечной слабости.
Четыре основных типа хронических или длительных воспалительных миопатий:
- полимиозит
- дерматомиозит
- миозит
- некротизирующая аутоиммунная миопатия
Причины
Причины миозита или общего мышечного воспаления:
- аутоиммунные нарушения, при которых нарушается иммунная система мышц;
- аллергическая реакция;
- вирус или другие инфекционные организмы, такие, например как бактерии.
Хотя причины множества воспалительных миопатий неизвестны, к основным относятся аутоиммунные нарушения.
Кто находится под угрозой?
Воспалительные миопатии встречаются достаточно редко. Данные заболевания могут поражать как взрослых, так и детей. Дерматомиозит является наиболее распространенной хронической формой у детей. Полимиозит и дерматомиозит чаще встречаются у женщин, в то время как миозит развивается главным образом у мужчин.
Признаки и симптомы
К общим симптомам хронической воспалительной миопатии относится медленная, но прогрессирующая мышечная дистрофия. Воспаление повреждает мышечные волокна, что вызывает слабость и может воздействовать на артерии и кровеносные сосуды, проходящие через мышцы. К другим симптомам относятся усталость после ходьбы, частые случаи нарушения равновесия или падения, а также трудности при глотании или дыхании. У некоторых людей может возникать боли в мышцах.
Полимиозит воздействует на скелетные мышцы (тип мышц, отвечающих за движения тела) по обе стороны тела. Заболевание редко встречается у людей моложе 20 лет. Обычно полимиозит развивается между 30 и 60 годами.
Признаки и симптомы полимиозита значительно различаются у отдельного больного, что может затруднить его диагностику. Прогрессирующая мышечная дистрофия, неконтролиуемая врачом, может привести к затруднению глотания, трудности при разговоре, принятия положения стоя или сидя, подъема по лестнице, перемещения предметов и т.д. У некоторых людей, страдающих полимиозитом, могут также развиваться артрит, одышка, сердечные аритмии (нерегулярные сердечные сокращения) или застойная сердечная недостаточность (когда сердце не справляется с перекачкой достаточное количество крови).
Дерматомиозит характеризуется кожной сыпью, которая предшествует или сопровождает прогрессирующую мышечную дистрофию. Сыпь, как правило, пятнистая, с фиолетовыми или красными пятнами, и появляется на веках и области локтей, колен и пальцев ног. Красные высыпания могут также возникать на лице, шее, плечах, верхней части туловища, спине и других местах. В подверженных сыпи областях может развиваться отек. Сыпь часто становится более отчетливой при воздействии солнца.
Взрослые люди, страдающие дерматомиозитом, могут терять вес или испытывать лихорадку со средней температурой 39-40°, воспаление легких, а также быть чувствительными к свету. Дерматомиозит у взрослых людей, в отличие от полимиозита, может сопровождаться отеком молочной железы, легких, женских половых органов или кишечника. У детей и взрослых с дерматомиозитом могут развиться отложения кальция, которые выражаются в виде твердых образований под кожей или мышцами (так называемый кальциноз). Кальциноз чаще всего встречается через один-три года после начала заболевания, но может возникать и много лет спустя. Эти отложения чаще встречаются при детском дерматомиозите, чем при дерматомиозите, который развивается у взрослых больных.
В отдельных случаях при полимиозите и дерматомиозите мышцы дистальных отделов тела, которые расположены вдали от центра тела, например, мышцы предплечий или вокруг лодыжек и запястий), могут подвергаться дистрофии при прогрессировании заболевания. Полимиозит и дерматомиозит могут быть вызваны коллагеновыми или аутоиммунными заболеваниями, такими как волчанка. Полимиозит также может быть связан с инфекционными заболеваниями, такими как ВИЧ.
Миозит с включениями (МСВ) является наиболее распространенной формой воспалительной миопатии у людей в возрасте 50 лет и старше и характеризуется медленной, прогрессирующей мышечной дистрофией. МСВ поражает как проксимальные, так и дистальные мышцы, как правило мышцы в бедрах и предплечьях, и часто наблюдается на обеих сторонам тела, хотя мышечная слабость может воздействовать только на одну сторону тела. Миозит с включениями также характеризуется дегенерацией мышц с многобелковыми агрегатами (прионами) в мышце, которые могут содержать токсины, наблюдаемые при болезни Альцгеймера и других нейродегенеративных заболеваниях.
Падения как правило являются первыми заметными симптомами. Заболевание часто начинается со слабости в запястьях и пальцах, что создает проблемы с захватом (например, застегиванием пуговицы) предметов. Больные могут испытывать слабость в мышцах запястья и пальца и атрофию (слабость или потеря мышечной массы) в мышцах предплечья и четырехглавой мышце бедра. Трудности при глотании возникают примерно в половине случаев МСВ при дистрофии мышц горла.
Симптомы заболевания обычно начинаются после 50 лет, хотя болезнь может развиваться и ранее. В отличие от полимиозита и дерматомиозита, миозит со включениями чаще встречается у мужчин, чем у женщин.
Некротизирующая аутоиммунная миопатия является редкой и относительно недавно признанной самостоятельной подгруппой воспалительных миопатий. Она может происходить в любом возрасте, но как правило затрагивает взрослое население. Его симптомы сходны с полимиозитом и дерматомиозитом, слабость как в верхней, так и в нижней части тела, трудности со вставанием со стула, подъем по лестнице или подъем предметов. Однако начало данных симптомов может быть более гнетущим и внезапным, достигая своего пика в течение нескольких дней или недель. К другим симптомам относятся усталость, потеря веса и боль в мышцах.
Некротизирующая аутоиммунная миопатия происходит как отдельно, так и совместно с вирусными инфекциями, вызванные заболеваниями соединительной ткани, например, такими как склеродермия, или, реже, приемом лекарственных средств, снижающих уровень холестерина (статины). Мышечная слабость и боль могут прогрессировать даже после того, как люди перестают принимать препараты.
Детские формы воспалительных миопатий имеют некоторое сходство со взрослым дерматомиозитом и полимиозитом. Они обычно касаются детей в возрасте от 2 до 15 лет. Симптомы включают проксимальную мышечную слабость и воспаление, отек (аномальную задержку жидкостей в тканях организма, вызывающую отек и припухлость), мышечную боль, утомление, кожную сыпь, боли в животе, лихорадку и контрактуры. Контрактуры суставов вызваны укорочением мышц или сухожилий вокруг суставов, воспалением в сухожилиях мышц и препятствуют свободному сокращению суставов.
У детей, страдающих воспалительными миопатиями, могут возникать проблемы при глотании и дыхании. Работа сердца также может нарушаться. У порядка от 20 до 40 процентов детей с ювенильным дерматомиозитом развивается кальциноз, который может вызвать существенную мышечную слабость и боль, суставную контрактуру, язвы кожи и пониженную мышечную массу.
Диагностика
Диагностика основана на изучении истории болезни, результатах физического обследования, которые включают тесты на мышечную силу и исследование образцов крови, которые показывают повышение уровня различных мышечных ферментов и аутоантител.
Диагностические инструменты включают в себя:
- электромиография для фиксации электрической активности, создаваемой мышцами во время сокращения и покоя
- УЗИ для отыскивания мышечного воспаления
- магнитно-резонансная томография, чтобы выявить аномальную мышечную анатомию.
Биопсийный образец мышечной ткани позволяет исследовать на предмет признаков хронического воспаления, потери мышечного волокна, деформаций сосудов или других изменений, характерных для диагностики определенного типа воспалительной миопатии. Биопсия кожи может показать видоизменения в коже, связанные с дерматомиозитом.
Лечение
Инъекции гена адренокортикотропного гормона могут быть еще одним вариантом лечения для людей, которые не реагируют на лечение либо не могут переносить другие медикаменты. Биологические методы лечения, такие как ингибиторы ритуксимаба или фактора некроза опухоли, такие как инфликсимаб или этанерцепт, могут быть использованы в тяжелых случаях, когда другие варианты лечения не помогли. Тем не менее, в настоящее время известно мало исследований, которые продемонстрировали, насколько хорошо эти средства лечат полимиозит и дерматомиозит.
Как правило физическая терапия рекомендуется для предотвращения атрофии мышц, а также для поддержания мышечной силы и диапазона движения. Следует избегать постельного режима в течение длительного периода времени, поскольку у людей может развиться атрофия мышц, снижение мышечной функции и суставные контрактуры. Диета с низким содержанием натрия может помочь уменьшить отеки и сердечно-сосудистые осложнения.
Большинству людей страдающих дерматомиозитом может быть полезным использование местной мази, содержащей кортикостероиды, такролимус или пимекролимус для лечения расстройства кожи. Таким людям рекомендуется использование защитного солнцезащитного крема и одежды, особенно тем больным, которые чувствительны к свету. В редких случаях может потребоваться хирургическое вмешательство для удаления отложений кальция, вызывающих неврологическую боль и рецидивирующие инфекции.
Не существует стандартного, основанного на научных доказательствах, курса лечения для миозита со включениями. Болезнь, как правило, не реагирует на прием кортикостероидов и иммунодепрессантов. Некоторые данные свидетельствуют о том, что иммунодепрессанты или внутривенный иммуноглобулин могут иметь небольшое, но кратковременное, благоприятное действие в небольшом числе случаев. Физическая терапия может оказывать полезное действие для поддержания мобильности. Остальная терапия является симптоматической и поддерживающей.
Прогноз
В большинстве случаев симптомы дерматомиозита контролируются при терапии. У людей с сердечно-сосудистыми заболеваниями болезнь как правило протекает в более тяжелой форме и более устойчива к терапии. Примерно одна треть людей, страдающих дерматомиозитом, восстанавливается после болезни, у одной трети заболевание носит рецидивирующий характер, а у другой третьей части людей происходит хроническое течение болезни.
Прогноз на полимиозит различен. Большинство людей довольно хорошо реагируют на терапию, но у некоторых людей болезнь развивается в тяжелой форме. Поскольку полимиозит может вызывать проблемы при глотании, больные могут испытывать недоедание. Они также подвергаются повышенному риску случаев падения, что может привести к переломам бедер и других костей, инвалидности или смерти. В редких случаях у людей с тяжелой и прогрессирующей формой мышечной дистрофии могут развиться дыхательная недостаточность или пневмония.
Хотя некротизирующая аутоиммунная миопатия хуже поддается лечению, чем полимиозит и дерматомиозит, она в целом хорошо реагирует на долгосрочные комбинации иммунодепрессантов.
МСВ, как правило, устойчива ко всем методам лечения, а имеющиеся в настоящее время методы лечения плохо замедляют ее прогрессирование.
ЛАБОРАТОРНЫЕ МЕТОДЫ ИССЛЕДОВАНИЯ ДЕТЕЙ ПЕРВЫХ 3 ЛЕТ ЖИЗНИ В НЕВРОЛОГИЧЕСКОЙ КЛИНИКЕЕЩЕ: НА ГЛАВНУЮ СТРАНИЦУ
nevrologvolgograd.ru
Идиопатические воспалительные миопатии > Клинические рекомендации РФ (Россия) > MedElement
Лечение ПМ/ДМ
Основные цели фармакотерапии ПМ/ДМ
· достижение полного клинического ответа (отсутствия клинико-лабораторной активности в течение, не менее чем 6 месяцев на фоне терапии) или ремиссии (отсутствия клинико-лабораторной активности в течение, не менее чем 6 месяцев на фоне отмены терапии) (уровень доказательности В),
· снижение риска комарбидных инфекций ГК) (уровень доказательности С)
· выявление и своевременное лечение пациентов с наибольшим риском ИПЛ
Общие рекомендации по лечению
· Лечение пациентов ПМ/ДМ должно проводиться врачами-ревматологами.
· В случае наличия ИПЛ с ФА при АСС – с привлечением пульмонологов и основываться на тесном взаимодействии врача и пациента
· Следует рекомендовать пациентам избегать факторов, которые могут спровоцировать обострение болезни: отказаться от пребывания на солнце, от курения, от контактов и инфекционными больными, избегать физических и психо-эмоциональных перегрузок.
· Следует рекомендовать пациентам исключить факторы, повышающие риск развития побочных эффектов терапии ГК: не употреблять в пищу сладкие продукты, включая мед и сладкие фрукты, повышающие риск развития стероидного сахарного диабета, также, исключение острой пищи, применение гастропротекторов с целью предотвращения язвенных осложнений (уровень доказательности С)
· Все пациенты нуждаются в активной профилактике и лечении глюкокортикоидного остеопороза. Подбор антиостеопоретической терапии зависит от результатов денситометрического исследования и оценки дополнительных факторов риска остеопороза (менопауза, эндокринные заболевания). В зависимости от исходных данных минеральной плотности костной ткани назначаются препараты кальция в сочетании с витамином Д, или эти же препараты в сочетании с бисфосфанатами.
· У пациентов ПМ/ДМ следует избегать внутримышечных инъекций, проведение которых, затрагивая мышечную ткань, может способствовать как формированию постинъекционных кальцинатов, так быть причиной ложноположительных результатов уровня креатинфосфокиназы (КФК).
Ведущая роль в лечении ПМ/ДМ отводится ГК. Основные принципы лечения ГК:
· Раннее начало терапии (в течение первых 3-х месяцев от начала симптомов) ассоциируется с благоприятным прогнозом.
· Адекватная инициальная доза: в зависимости от тяжести заболевания начальная доза колеблется от 1 до 2 мг/кг/сут.
· Ежедневный прием ГК.
· Суточную дозу ГК в начале лечения следует делить на 3 приема (оценивая ее переносимость), однако в течение первой половины дня; затем перевести пациента на прием полной дозы ГК в утренние часы.
· Оценка эффективности терапии проводиться через 2-4 недели от начала терапии ГК. Положительный эффект терапии расценивается при начавшемся снижении уровня КФК, АСТ, АЛТ, уменьшении интенсивности кожных проявлений, нарастании мышечной силы.
Отсутствие положительной динамики в течение 4 недель требует повторного проведения дифференциального диагноза с фенотипически схожими нозологиями, включающими в клинической картине миопатический синдромам, в т.ч., пересмотра морфологического материала.
· Длительность инициальной дозы ГК составляет, в среднем, 2,5-3 месяца.
· Снижение дозы ГК начинается при нормализации уровня КФК в сыворотке крови, исчезновении спонтанной активности при и-ЭМГ, нарастании мышечной силы, объема движений и проводиться под строгим клинико-лабораторным контролем. Доза ГК постепенно снижается по ¼ дозы от исходной в месяц, в среднем, по ½ — ¼ таблетки в 5-7-10 дней до достижения поддерживающего уровня. Темп снижения зависит от исходной дозы ГК и степени активности болезни. Чем ниже доза ГК, тем медленнее ее снижение.
· Поддерживающая доза ГК индивидуальна: 5-10, реже 15 мг/сутки и зависит от клинико-иммунологического подтипа болезни, возраста больного. При ЮДМ известны случаи клинико-лабораторной ремиссии на фоне длительной отмены терапии. Полная отмена ГК у взрослых пациентов, как правило, ведет к обострению болезни, даже если они несколько лет находились в состоянии полного клинического ответа.
Пульс-терапия ГК у взрослых пациентов не является основополагающей при ПМ/ДМ и не служит поводом для применения меньших (не адекватных) доз ГК назначаемых внутрь, как в острый период болезни, так и при ее обострении.
Потенциальные показания к подключению иммуносупрессивной терапии
· Принадлежность больных к клинико-иммунологическим подтипам ПМ/ДМ, особенностью которых является заведомо «плохой ответ» на терапию ГК: АСС c ФА, у пациентов антител к SRP
· Язвенно-некротический васкулит
· Обострение заболевания при снижении дозы ГК
· Стероидрезистентность у больных, ране получавших неадекватно малые дозы ГК
· Неэффективность ГК в течение 3-х месяцев
· Тяжелые побочные эффекты ГК, лимитирующие назначение адекватной дозы ГК (неконтролируемые сахарный диабет или артериальная гипертензия, острая язва желудка, множественные остеопоретические переломы)
Рекомендации по лечению ПМ/ДМ соответственно наиболее тяжелым синдромам ИПЛ с синдромом ФА при АСС
· Наиболее тяжелым и плохо контролируемым монотерапией ГК при ПМ/ДМ синдромом является АСС. Плохой прогноз определяется вовлечением в патологический процесс легочной ткани — ИПЛ с развитием ФА.
· Объем терапии и выбор препарата (в сочетании с ГК) определяется тяжестью ИПЛ (по данным КТ и ФЛТ: форсированной ЖЕЛ, DLCO и с учетом анамнеза (ранее применяемые иммуносупрессивные препараты).
· Основное место в лечении ИПЛ занимает ЦФ, назначаемый внутривенно в дозе 500 мг/м2 -750 мг/м2 мг в месяц в сочетании с ГК (уровень доказательности А)
· Длительность ЦФ должна быть не менее 6 месяцев (уровень доказательности С)
· Контроль эффективности ЦФ осуществляется по динамической оценке (1 раз в 6 месяцев) форсированной ЖЕЛ, показателей DLCO (уровень доказательности А), а также данных КТВР легких.
· При агрессивном течении СФА при выраженном снижении ЖЕЛ и DLCO, а также, в случае неэффективности ранее применяемой терапии ЦФ, целесообразно применение РТМ.
· Применение ММФ рассматривается в качестве терапии «второго» ряда при ИПЛ в случае невозможности применения ЦФ или РТМ
Дисфагия
· Дисфагия является фактором риска аспирационной пневмонии, течение и терапия которой осложняется иммуноскомпроментированностью пациентов, связанной с терапией высокими дозами ГК и цитостатиков.
· Рекомендовано проведение пульс-терапии ГК (метипред 1000мг) N 3 в сочетании с пероральным приемом ГК в адекватной дозе.
· Тяжелая дисфагия является потенциальным показанием ВВИГ.
Наличие дисфагии у больных ПМ/ДМ служит поводом для проведения более активного онкопоиска (уровень доказательности Д).
Язвенно-некротический васкулит
Наличие язвенно-некротического васкулита является показанием для проведения пульс-терапии циклофосфамидом в дозе 600-800-1000 мг в месяц в сочетании метилпреднизолоном 500-1000мг.
Кожный синдром при ДМ в сочетании с проксимальной мышечной слабостью отражает активность болезни и, как правило, контролируется ГК в адекватных дозах в острый период болезни.
При резистентном кожном синдроме, сохраняющемся на фоне восстановления мышечной силы, рекомендуется применение антималярийных препаратов (гидроксихлорохин по 200–400 мг/сут), ММФ, топических стероидов.
Наличие резистентного кожного синдрома и/или язвенно-некротического васкулита у больных ПМ/ДМ служит поводом для проведения более активного онкопоиска (уровень доказательности С).
Лихорадка или субфебрилитет встречаются редко, главным образом при АСС с острым началам болезни.
· Контролируется ГК и не требует дополнительной терапии (уровень доказательности В).
При появлении субфебрилитета (или лихорадки) у пациентов на фоне лечения ГК в период клинико-лабораторной положительной динамики – исключение присоединения сопутствующей инфекции. Необходимо учитывать атипизм течения инфекционных осложнений на фоне иммуносупрессивной терапии.
Поражение суставов
· Наличие артрита при ПМ/ДМ может присутствовать в начале болезни. Артриты входят в состав симптомокомплекса АСС, хорошо контролируются ГК и не требуют дополнительного лечения.
· Сгибательные контрактуры, как правило, локтевых, реже коленных суставов, развиваются в острый период ПМ/ДМ и обусловлены воспалительным поражением мышечной ткани, а не непосредственным поражением суставов. Дополнительного медикаментозного лечения не требуется (уровень доказательности С).
Кальциноз мягких тканей
· Кальциноз мягких тканей наиболее часто присутствует (и более агрессивен) при ЮДМ
· Появление множественных кальцинатов, как правило, сопутствует острому течению ПМ/ДМ. Кальцинаты сохраняются на фоне снижения активности болезни, даже при достижении клинико-лабораторной ремиссии и наиболее выражении при ЮДМ.
· При ЮДМ, с целью снижения риска развития кальциноза и его распространенности применяется пульс-терапия ГК в дозе 1-2 мг/кг/сут.
· Хирургическое лечение малоэффективно, поскольку повышает риск присоединения вторичной инфекции и может спровоцировать появление новых кальцинатов.
· В качестве медикаментозной терапии применяютт бисфосфонаты (ксидифон, фосамакс, фосаванс и др.), однако полного контроля над процессом гетеротопического кальцийобразования не достигается.
· Для лечения кальциноза применяется, также динатриевая соль этилендиаминотетрауксусной кислоты (Na2ЭДТА), образующей комплексные соединения с различными катионами, в т.ч.с ионами Са2+ и способствует выделению их с мочой.
· Имеются данные об эффективном предотвращении прогрессирования кальциноза при применении ВВИГ в течение 2 дней каждый месяц в сочетании с метилпреднизолоном (уровень доказательности С).
Традиционные иммуносупрессивные препараты, применяемые в лечении ПМ/ДМ
· Метотрексат по 7,5–25 мг/нед внутрь или внутривенно (при недостаточной эффективности или плохой переносимости перорального приема препарата, особенно в высоких дозах).
· Азатиоприн по 2–3 мг/кг/сут (100–200 мг/сут)
· Циклоспорин А по 2,5–5.,0 мг/кг/cутки назначают пациентам с резистентными к ГК формами заболевания, в т.ч. при хроническом течении болезни, связанной с неадекватно малой инициальной дозой ГК (уровень доказательности С).
· ММФ. Имеются данные об эффективности ММФ при ИПЛ и резистентном кожном синдроме. Прием начинают с дозы 1000 мг/сут (в 2 приема), постепенно титруя дозу до 2000 мг/сут под контролем показателей общего и биохимического анализов крови (уровень доказательности С).
Общие принципы лечения иммуносупрессивными препаратами:
— титрование дозы: назначение с небольшой дозы и постепенное ее повышение под контролем переносимости.
— контроль переносимости: оценка уровня гемоглобина, числа лейкоцитов, тромбоцитов, азота мочевины, креатинина, активности АСТ, АЛТ. При уменьшении числа лейкоцитов менее 2,5х109/л и/или тромбоцитов – менее 100 х 109/л и повышении концентрации АСТ, АЛТ более чем в 3 раза от верхней границы нормы, лечение необходимо прекратить до устранения симптомов токсичности.
— при присоединении интеркурренотой инфекции, в т.ч. герпетической – временная отмена иммуносупрессивных препаратов до исчезновения ее признаков.
Применение ВВИГ 2 г/кг 1 раз в месяц в течение 3 месяцев является эффективным методом лечения ПМ/ДМ (особенно ЮДМ), резистентного к стандартной терапии. Потенциальным показанием для ВВИГ является тяжелая дисфагия. (уровень доказательности В)
Плазмаферез следует использовать главным образом у больных с тяжёлым, резистентным к другим метода лечения ПМ/ДМ в сочетании с ГК и цитотоксическими препаратами.
Новые направления терапии ПМ/ДМ. Биологические препараты
В настоящее время активно изучается роль и место биологической терапии в терапии ПМ/ДМ.
— Применение в терапии ПМ/ДМ ингибиторов фактора некроза опухоли α TNF-a (инфликсимаба) не принесло желаемых результатов: поскольку он не способен контролировать активность болезни, в том числе ИПЛ, а также, увеличивают риск оппортунистических инфекций [10, 44].
— Имеются данные об успешном применении этанерцепта в качестве стероидсберегающей терапии. (Уровень доказательности Д)
— Применение блокаторов ко-стимуляции Т-лимфоцитов (абатацепта) в сочетании с тиосульфат натрия при ЮДМ с язвенно-некротичнским васкулитом и прогрессирующим кальцинозом оказало положительный эффект в виде нарастания мышечной силы, восстановления целостности кожный покровов, снижения прогрессирования кальциноза, что позволило снизить поддерживающую дозу ГК. (Уровень доказательности Д)
— Особое место среди биологических препаратов, на сегодняшний день, применяемых при ПМ/ДМ, занимает использование анти В-клеточной терапии. Накоплен положительный опыт по применению РТМ у пациентов с тяжелым мышечным поражением и при АСС с СФА, резистентных к ГК и применяемой ранее традиционной цитостатической терапии (уровень доказательности Д)
Практически все авторы описывают высокую эффективность РТМ при ПМ/ДМ. Так, на фоне терапии РТМ (в сочетании с ГК) наблюдается положительная клинико-лабораторная динамика (уменьшение выраженности кожного синдрома, нарастание мышечной силы).
— В случае применения РТМ при АСС с СФА, позитивный эффект наблюдался более, чем у 70% больных в виде увеличения показателей функции внешнего дыхания: увеличения показателей ЖЕЛ и DLCO, а также уменьшения инфильтратов по КТ грудной клетки.
— Максимальный эффект развивался через 12 недель после первой инфузии и коррелировал со снижением CD 20 + В клеток.
Ведение пациентов ПМ/ДМ с хроническим течением болезни, связанным с неадекватно малой инициальной дозой ГК
· Сложность ведения таких пациентов обусловлена развитием поствоспалительной фиброзной и жировой инволюции мышечной ткани (при назначении не адекватной инициальной дозы ГК). В этом случае отсутствует (или минимален) воспалительный компонент (миозит), являющийся субстратом для проведения противовоспалительной терапии ГК.
· Клинически — сохраняется проксимальная мышечная слабость, однако показатели активности болезни (уровень КФК, данные и-ЭМГ, биоптата мышечной ткани) не свидетельствуют в пользу текущего воспалительного процесса.
· Присутствие фиброзной и жировой инволюции мышечной ткани подтверждается при МРТ исследовании проксимальных отделов конечностей.
· Повышение дозы ГК целесообразно при наличии, хотя бы минимальных, признаков воспаления мышечной ткани.
· Хроническое течение болезни, связанное с неадекватно малой инициальной дозой ГК является потенциальным показанием для подключения иммуносупрессивной терапии (Циклоспорин А, ММФ, метотрексат, азатиоприн) .
Реабилитационные мероприятия и обучение пациентов.
Проводятся в зависимости от стадии заболевания
· В острую фазу противопоказаны ЛФК и физические нагрузки, проводимые пациентами «через силу»; допускаются только пассивные упражнения
· В стадию выздоровления — изометрические, а затем изотонические упражнения
· В хронической стадии — анаэробные упражнения
Профилактика ГК- остеопороза
Препараты кальция в сочетание с витамином Д3, бисфосфанаты.
Профилактика язвенных осложнений
Гастропротекторы (миозпростол, ранитидин, омепразол).
Профилактика стероидного диабета
Строгое исключение потребления продуктов, содержащих глюкозу, в т.ч., сладких фруктов, соков и йогуртов.
Предосторожности: Исключение контакта с инфекционными больными – во избежание присоединения вторичной инфекции.
Избегание физических перегрузок (в острый период ЛФК противопоказана, только пассивные движения).
diseases.medelement.com
Идиопатические воспалительные миопатии | Антелава О.А., Хитров А.Н., Насонов Е.Л.
Воспалительные заболевания мышц – группа заболеваний, основным проявлением которых является мышечная слабость, связанная с воспалением поперечно–полосатой мускулатуры [1]. К ним относятся: идиопатические воспалительные миопатии (ИВМ), миопатии, связанные с инфекцией, и миопатии, связанные с воздействием лекарственных препаратов и токсинов. Наиболее яркими представителями ИВМ являются системные аутоиммунные ревматические заболевания неизвестной этиологии: полимиозит (ПМ) и дерматомиозит (ДМ). Наряду с ПМ и ДМ в группу ИВМ входят: ювенильный ДМ, миозит, ассоциирующийся с СЗСТ (перекрестный синдром), миозит, ассоциирующийся с опухолями, миозит с внутриклеточными включениями и несколько более редких заболеваний (оссифицирующий миозит, локализованный или очаговый миозит, гигантоклеточный миозит, эозинофильный миозит) (по Wortman R.L. 1994) [1].
История вопроса. Первые описания дерматомиозита и полимиозита принадлежат немецким клиницистам в период между 1886 и 1891 г. Термин «полимиозит» предложен E. Wagner в 1886 году, а в 1891 г. – H. Unverricht обратил внимание на сочетание кожного и мышечного поражения и использовал термин «дерматомиозит». Около 2/3 описаний XIX века соответствуют полимиозиту, оставшиеся – дерматомиозиту. В 1916 году описана ассоциация со злокачественными опухолями, однако причинная взаимосвязь предположена только в 1935 году. В 1967 Chou впервые описал миозит с включениями, хотя сам термин «миозит с включениями» введен Yunis и Samaha в 1971 году, когда в гистопатологических срезах мышечной ткани были обнаружены характерные ядерные цитоплазматические включения [2].
Частота ПМ/ДМ в популяции колеблется от 2 до 10 на 1 млн населения в год. ДМ, реже ПМ, ассоциирующиеся с опухолями, составляют примерно 20% от всех случаев воспалительных миопатий. Опухоли могут развиваться до появления признаков ИВМ, одновременно с ними или после их появления. Частота злокачественных новообразований при ПМ/ДМ в 12 раз выше, чем в популяции. На фоне злокачественных новообразований чаще развивается ДМ, чем ПМ [1,3].
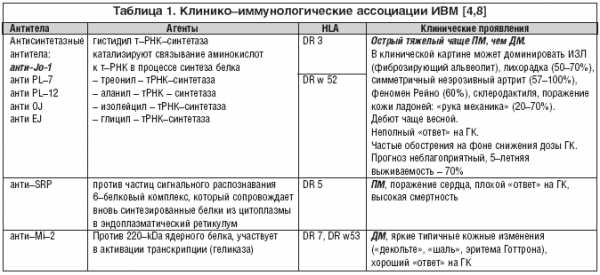
Этиология. На роль генетической предрасположенности указывает возможность развития ПМ/ДМ у монозиготных близнецов и кровных родственников пациентов. Носительство некоторых HLA (HLA–В8/DR3, HLA B14 и HLA B40) более тесно связано не с самим заболеванием, а с определенными иммунологическими нарушениями, в первую очередь гиперпродукцией миозит–специфических аутоантител (табл. 1) [1].
Патогенез. В основе патогенеза ПМ/ДМ лежат клеточные и гуморальные иммунные реакции. Следует отметить, что в патогенезе ПМ и ДМ существуют принципиальные иммунопатологические различия.
При ДМ развивается гуморальный иммунный ответ, приводящий к активации системы комплемента и формированию активированного С3, что ведет к формированию C3b неоантигена и мембранолитического атакующего комплекса, который откладывается в эндотелиальных клетках и вокруг них, в стенке эндомизиальных капилляров. Отложение мембранолитического атакующего комплекса ведет к деструкции и уменьшению числа капилляров с ишемией и микроинфарктами миоцитов, наиболее выраженного на периферии пучка. В результате развивается картина малого числа увеличенных в диаметре капилляров и перифасцикулярная атрофия [3].
При ДМ также наблюдается миграция В–клеток, CD4+ Т–лимфоцитов и макрофагов, образующих внутримышечный инфильтрат. Миграции способствуют васкулярные молекулы клеточной адгезии 1 (VCAM1) и межклеточные молекулы адгезии 1 (ICAM1), экспрессия которых на эндотелиальных клетках повышена за счет воздействия высвобожденных цитокинов, с последующим поражением внутримышечных микрососудов и развитием внутримышечной васкулопатии. Цитокины, высвобожденные активированными Т– и В–клетками, усиливают процесс трансмиграции воспалительных клеток в мышцу [3].
При ПМ и миозите с включениями увеличивается клон аутоинвазивных СD8+ Т–клеток. CD8+ Т–лимфоциты синтезируют цитотоксические субстанции (перфорин, гранзим) и через перфориновый путь оказывают миотоксическое действие на миофибриллы, экспрессирующие молекулы HLA–I класса [1–4]. В дебюте патогенетического процесса ПМ и миозита с включениями наблюдается повышение экспрессии молекул HLA–I на мышечных волокнах, даже в отсутствие аутоинвазивных СD8+ Т–клеток [3].
Наиболее часто болезнь дебютирует недомоганием, общей слабостью, миалгиями, преходящим симметричным поражением суставов, поражением кожи. Началу болезни может предшествовать инсоляция. Затем, в течение нескольких недель (месяцев), постепенно нарастает слабость проксимальных групп мышц. У детей и лиц молодого возраста может наблюдаться более острое начало, часто сочетающееся с выраженными конституциональными проявлениями (лихорадка, похудание и др.). У больных с амиопатическим ДМ в течение длительного времени может наблюдаться типичное для ДМ поражение кожи при отсутствии мышечной слабости [1].
Ведущим клиническим признаком заболевания [5,6] является поражение мышц, выражающееся симметричной слабостью проксимальных групп мышц верхних и нижних конечностей и мышц шеи, ведущей к затруднению при подъеме с низкого стула, посадке в транспорт, при умывании и причесывании; больной не может оторвать голову от подушки. Наблюдаются: изменение походки и эпизоды неожиданных падений. Может развиваться отек мышц. Характерно поражение мышц глотки, гортани и верхней трети пищевода (дисфония, дисфагия). Амиотрофии развиваются только у больных, длительно страдающих ПМ/ДМ.
Поражение дистальной мускулатуры не характерно и наблюдается, главным образом, при миозите с «включениями».
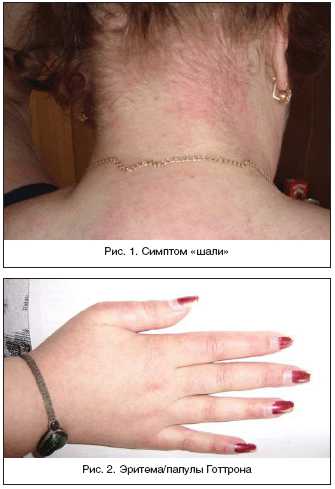
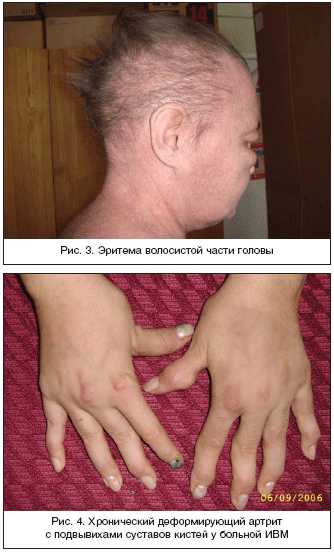
Поражение кожи при ДМ часто предшествует развитию мышечной слабости. Характерным признаком является эритематозная («гелиотропная») сыпь, локализующаяся на верхних веках, скулах, в зоне «декольте» и «шали» (рис. 1), над локтевыми, коленными, пястнофаланговыми и проксимальными межфаланговыми суставами – эритема/папулы Готтрона (рис. 2), эритема волосистой части головы (рис. 3). Также наблюдаются шелушение и трещины на коже ладоней («рука механика»), околоногтевая эритема, фотодерматит, кожный зуд. Могут наблюдаться такие формы сосудистой патологии, как инфаркты околоногтевого ложа, петехии и сетчатое ливедо [6,7].
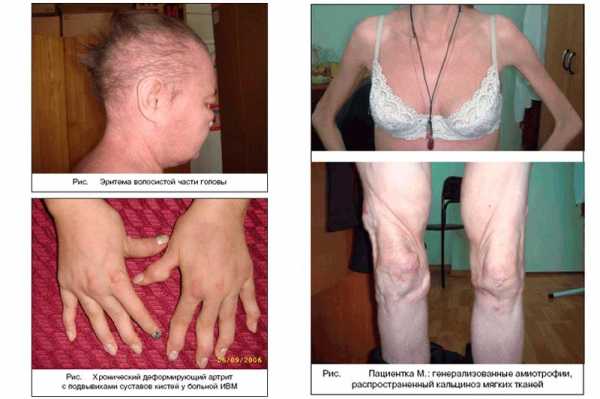
Артриты/артралгии наблюдаются достаточно редко и быстро купируются при назначении глюкокортикоидов (ГК). Развитие хронического деформирующего артрита с подвывихами суставов кистей наблюдается редко и не сопровождается эрозивными изменениями (синдром Жаку) (рис. 4) [1].
Кальциноз мягких тканей наиболее часто наблюдается при ЮДМ, при перекрестных синдромах миозита (например, с системной склеродермией) или на поздних стадиях ПМ/ДМ. Кальцинаты располагаются подкожно, в соединительной ткани, вокруг мышечных волокон и, наиболее часто, в зонах травматизации или после внутримышечных инъекций, например, на ягодицах [1].
Наиболее часто встречающимся органным поражением является поражение дыхательной системы (80%) [8,9]. При ПМ/ДМ наблюдаются различные варианты поражения дыхательной системы.
У подавляющего большинства пациентов наблюдается вовлечение в патологический процесс как межреберных мышц, так и диафрагмы. Высокое стояние куполов диафрагмы и вялость ее дыхательной экскурсии приводит к экспираторной одышке, изменению функции внешнего дыхания по рестриктивному типу. Кроме того, нарушение глотания, связанное с поражением глоточных мышц, может приводить к аспирации пищи и слюны с последующим развитием аспирационной пневмонии.
Наиболее частой формой легочного поражения является пневмония, которая встречается при ПМ/ДМ в 29–54% случаев. Ведущую роль в ее развитии играет аспирация пищи. Риск развития пневмоний и трудности при их лечении возрастают в связи выраженным иммунодефицитом пациентов с ПМ/ДМ, обусловленным длительным приемом высоких доз ГК и иммуносупрессивной терапии. Кроме того, гиповентиляционный синдром усугубляет риск развития пневмонии [8].
Наиболее тяжелым поражением легочной ткани является фиброзирующий альвеолит, который в зарубежной литературе принято называть интерстициальным заболеванием легких (ИЗЛ). Фиброзирующий альвеолит при ПМ/ДМ имеет широкий спектр клинико–лабораторных проявлений и сходен с идиопатическим фиброзирующим альвеолитом. Targoff I.N. c соавт.1992, выделяют три формы синдрома фиброзирующего альвеолита: 1 – По типу быстро прогрессирующего синдрома фиброзирующего альвеолита (синдром Хаммана–Ричи): острый непродуктивный кашель, одышка в покое, лихорадка. На рентгенограммах – множественные мелкоочаговые затемнения и сетчатая деформация легочного рисунка. Симптомы поражения мускулатуры могут быть на втором плане. Прогноз в данном случае, неблагоприятный. 2 – Более медленное развитие болезни, в дебюте: одышка при физической нагрузке и/или непродуктивный кашель. Клинические проявления могут предшествовать мышечному поражению, возникать одновременно с ним или развиваться на фоне имеющегося тяжелого миозита. У ряда больных 1 и 2 групп может присутствовать тахипноэ, крепитация в нижних отделах легких, реже развитие легочного сердца. 3 – Субклиническое поражение: яркая клиническая легочная симптоматика отсутствует. Интерстициальные изменения в легочной ткани выявляются только при помощи специальных методов исследования (КТ, рентгенография) [8].
Так, клиника фиброзирующего альвеолита может опережать развитие мышечного синдрома или проявляться одновременно с ним. Однако наиболее часто клинические признаки синдрома фиброзирующего альвеолита развиваются на фоне клинической картины ПМ/ДМ, как правило, на ранних этапах болезни.
Поражение сердца в большинстве случаев протекает без явных клинических признаков повреждения. Ведущее место в структуре кардиологической патологии при ИВМ занимает поражение миокарда, наиболее часто проявляющееся нарушением ритма (суправентрикулярная или желудочковая экстрасистолия) и проводимости (наиболее часто – блокада левой ножки пучка Гиса), а также снижением его сократительной способности. Наиболее частым проявлением нарушения сократительной функции сердца является снижение локальной сократимости в области задней стенки левого желудочка и межжелудочковой перегородки. Крайне редко развиваются миокардит или застойная сердечная недостаточность на фоне миокардиофиброза [10].
Поражение ЖКТ: нарушение глотания обусловлено поражением мышц глотки, гипотония пищевода (верхняя треть) [1]. Поражение почек наблюдается крайне редко, как правило, при overlap–синдромах, в основном с ССД. Возможна протеинурия. Описаны единичные случаи развития рабдомиолиза (миоглобинурия – острая почечная недостаточность) [1,4].
Феномен Рейно чаще наблюдается при ДМ, антисинтетазном синдроме и у больных с перекрестным синдромом ПМ/ДМ и со смешанным заболеванием соединительной ткани [1,4,11].
Клинико–иммунологические ассоциации (табл. 1). Согласно современным представлениям, развитие ИВМ связано с синтезом целого «семейства» аутоантител, направленных к цитоплазматическим белкам и рибонуклеиновым кислотам. Эти антитела присутствуют в сыворотках у почти 90% больных, рассматриваются как миозитспецифические антитела и подразделяются на подгруппы [4,8]. Выявление аутоантител каждой подгруппы ассоциировано с определенным симптомокомплексом. Наиболее тяжелым является «антисинтетазный» синдром, который маркируют антитела к аминоацилсинтетазам тРНК (Jo–1и др.) [11].
Лабораторные данные включают повышение уровня сывороточной креатинфосфокиназы (КФК), лактатдегидрогеназы (ЛДГ), аспарагиновой (АСТ), аланиновой (АЛТ) трансаминаз. При мышечном повреждении может наблюдаться снижение уровня сывороточного креатинина [11–13].
АНФ определяется у 50–80%, чаще у больных с перекрестными синдромами миозита с другими СЗСТ и в более высоких титрах [14]. Определение антител к аминоацилсинтетазам транспортной РНК, в первую очередь Jo–1: диагностический критерий ПМ/ДМ и лабораторный маркер «антисинтазного» синдрома [15–18].
Электромиографическое исследование игольчатыми электродами (и–ЭМГ) проводится с целью подтверждения первично–мышечного поражения, определения степени активности воспалительного процесса и некроза мышечных волокон. При ПМ/ДМ наблюдается снижение амплитуды и средней длительности потенциалов двигательных единиц (ПДЕ), полифазия, псевдополифазия и спонтанная активность (СА) мышечных волокон (потенциалы фибрилляций и положительные острые волны). Поведение и–ЭМГ необходимо и в динамике, для оценки эффективности проводимой терапии [19–20].
Согласно классическим диагностическим критериям, в мышечном биоптате выявляются: некроз и атрофия мышечных волокон, выраженная лимфогистиоцитарная инфильтрация, умеренная регенерация мышечных волокон, потеря поперечнополосатой исчерченности [12,24].
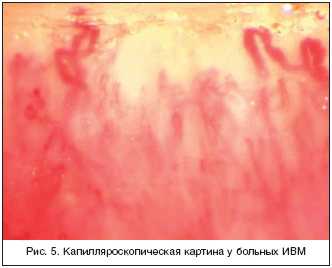
Капилляроскопическая картина представлена деструкцией и дезорганизацией капилляров, уменьшением их числа, увеличением размера; неоангиогенезом, а также формированием так называемых «кустовидных» капилляров (рис. 5).
Рентгенологическое исследование легких или рентгеновская компьютерная томография с высоким разрешением способствует выявлению патологических изменений легочной ткани, от базального пневмосклероза до острого фиброзирующего альвеолита [8].
В последние годы многие исследователи опираются на результаты магнитно–резонансной томографии (МРТ) [22]. МРТ позволяет провести раннюю диагностику заболевания благодаря выявлению отека мышечной ткани даже до клинических проявлений поражения мышц, особенно в случае дерматомиозита, когда поражение кожных покровов уже очевидно. Отек мышечной ткани является индикатором активности болезни.
За годы изучения ИВМ неоднократно повторялись попытки разработки оптимальных диагностических критериев ИВМ: Bohan and Peter 1975; Dalakas 1991; Griggs 1995; Tanimoto 1995; Targoff 1997; Mastaglia 2002; Van der Meulen 2003; Dalakas, Hohlfeld 2003 [5–13]. Наиболее широко в настоящее время используются критерии Bohan and Peter 1975 и Tanimoto 1995 (табл. 2) [12,13,23–28].
Вызывают интерес диагностические критерии, предложенные Dalakas в 1991 году, которые основаны на специфических иммуногистопатологических особенностях мышечного инфильтрата и позволяют дифференцировать ДМ и ПМ от других миопатических синдромов (см. дифференциальная диагностика). Так, по мнению Dalakas, мышечный биоптат при достоверном ПМ имеет инвазию CD8+ лимфоцитами ненекротизированных мышечных волокон, экспрессирующих HLA–I класса гистосовместимости. При этом отсутствуют вакуоли, характерные для миозита с «включениями». Достоверный диагноз ДМ по Dalakas характеризуется наличием перифасциальной атрофии и воспалительных инфильтратов в мышцах в сочетании с характерными кожными изменениями [21].
Дифференциальная диагностика [19–20,29] ИВМ представляет сложную проблему и проводится с широким спектром заболеваний, сопровождающихся также мышечной слабостью, увеличением КФК и первично–мышечными электромиографическими изменениями.
Истинную проксимальную мышечную слабость следует отличать от общей слабости, утомляемости или миалгии, вызываемыми различными причинами.
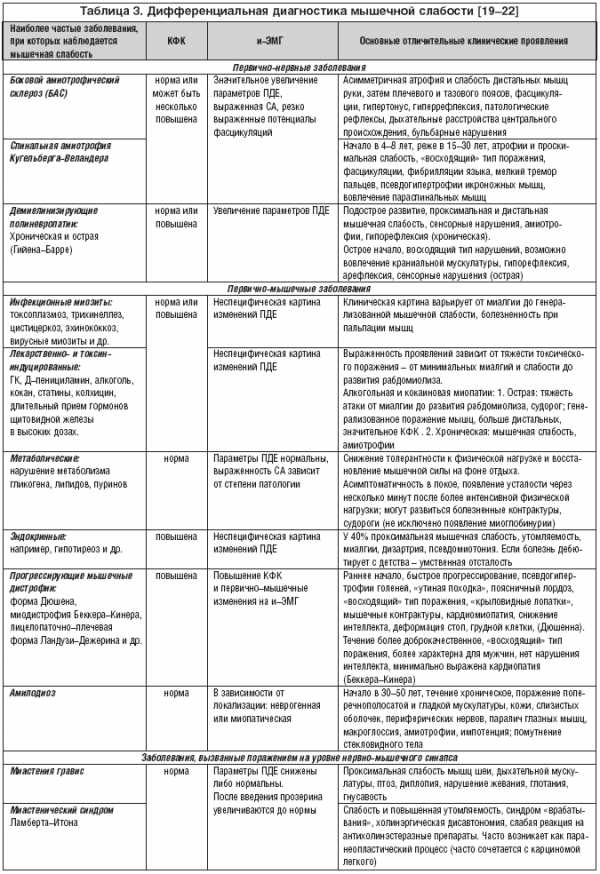
Учитывая актуальность и сложность проблемы дифференциальной диагностики воспалительных миопатий, еще в 1975 году Bohan and Peter были выделены критерии исключения [12], объединяющие различные нозологическое формы, где одним из клинических проявлений может являться мышечная слабость. В первую очередь, это неврологические заболевания, миастения гравис и миодистрофии, а также саркоидоз, инфекционные болезни (трихенеллез, цистеркоз, токсоплазмоз), токсин– и лекарственно–индуцированные миопатии, рабдомиолиз, метаболические миопатии (например, болезнь Мак Ардла) и эндокринопатии (табл. 3).
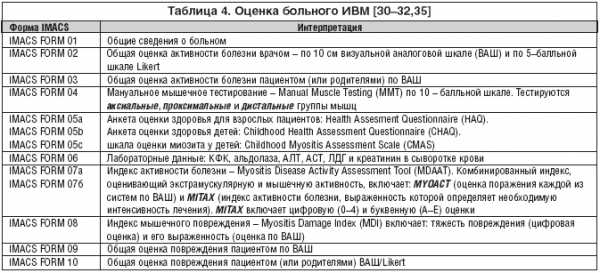
В 2000 г. создана международная мультидисциплинарная группа исследователей по оценке миозита и клинических исследований (International Myositis Assessment and Clinical Studies Group – IMACS) под руководством профессора P. Plotz. Разработаны стандартизированные методы обследования больных ИВМ (табл. 4) [30–32].
Предложены следующие дефиниции:
Активность рассматривается как частично или полностью обратимые явления, связанные непосредственно с воспалительным процессом в мышцах. Повреждение – критерий исхода болезни, отражающий поствоспалительные необратимые изменения в мышцах и других органах (рубцевание, фиброз, амиотрофии, замещение жировой тканью) и в результате проводимой терапии (стероидная катаракта, сахарный диабет, инфекционные осложнения и др.) [30–34]. Полный клинический ответ на терапию определяется как отсутствие признаков активности болезни на фоне продолжения проводимого лечения, а полная клиническая ремиссия – как отсутствие признаков активности миозита на фоне отсутствия любой терапии в течение 6 месяцев [32].
Основу лечения ИВМ составляют ГК [36]. Раннее начало терапии (в течение первых 3–х месяцев от начала симптомов) ассоциируется с благоприятным прогнозом [37,38]. В зависимости от тяжести заболевания начальная доза колеблется от 1 до 2 мг/кг/сут. Улучшение состояния (нарастание мышечной силы) больных ПМ/ДМ развивается медленнее, чем при других ревматических заболеваниях. При отсутствии положительной динамики в течение 4 недель следует увеличить дозу ГК. После достижения эффекта или значительного улучшения (нарастание мышечной силы и КФК) дозу ГК постепенно снижают до поддерживающей. Снижение дозы ГК должно проводиться под строгим клинико–лабораторным контролем [39].
Пульс–терапия ГК редко эффективна, применяется, главным образом, при ЮДМ, когда она может предотвратить быстрое прогрессирование миопатии и развитие кальциноза. При ПМ/ДМ у взрослых пульс–терапию ГК применяют в случае быстрого прогрессирования дисфагии (риск аспирационной пневмонии) и развития системных проявлений (миокардит, альвеолит).
При наличии факторов риска неблагоприятного прогноза (позднее назначение ГК–терапии, тяжелая мышечная слабость, наличие дисфагии) [37,38], при невозможности назначения адекватной дозы ГК из–за побочных эффектов или при недостаточной эффективности ГК применяются препараты «второго» ряда: метотрексат, азатиоприн [37], циклофосфамид: («препарат выбора» при интерстициальном легочном фиброзе), циклоспорин А.
Имеются данные о положительном эффекте циклоспорина А в отношении прогрессирования интерстициального легочного фиброза [40,41]. Накоплен некоторый опыт лечения такролимусом, который сходен по иммуномодулирующим эффектам с циклоспорином А [39,42,43].
Одним из наиболее обнадеживающих направлений фармакотерапии ИВМ многие годы считается использование внутривенного иммуноглобулина. Еще в 1993 г. М.С. Dalakas [44] доказан его положительный клинико–лабораторный эффект. Однако кратковременность эффекта после прекращения его применения требует проведения повторных инфузий. Потенциальным показанием для внутривенного иммуноглобулина является тяжелая дисфагия [42].
Относительно новым цитостатическим препаратом для лечения аутоиммунных заболеваний является микофенолата мофетил – иммунодепрессант антиметаболического типа, обладающий хорошей переносимостью согласно литературным данным [42,43].
В последние годы для лечения ИВМ все чаще применяются ингибиторы фактора некроза опухоли? (ФНО–a), в частности инфликсимаб [42,45–48]. Появились данные об успешном применении при аутоиммунных заболеваниях препаратов, блокирующих пролиферацию В–клеток, одним из которых является ритуксимаб. Имеются отдельные сообщения об его эффективности при ДМ [42,49,50].
При резистентном кожном синдроме, сохраняющемся на фоне восстановления мышечной силы, рекомендуется применение антималярийных препаратов, топических стероидов, такролимуса, микофенолат мофетила [39,42,43].
Реабилитационные мероприятия проводятся в зависимости от стадии заболевания. В острую фазу показаны пассивные упражнения, в стадию выздоровления – изометрические, а затем изотонические упражнения и, наконец, в хронической стадии – анаэробные упражнения [1].
Прогноз. Внедрение в клиническую практику ГК существенно увеличило выживаемость больных ПМ/ДМ, которая составляет 90% через 5 лет после постановки диагноза, за исключением больных онкомиозитом. Факторами, ассоциирующимися с неблагоприятным прогнозом при ПМ/ДМ, являются пожилой возраст, поздно поставленный диагноз, неадекватная терапия ГК в начале болезни, тяжелое течение миозита, антисинтетазный синдром [1,8].

Литература
1. Насонов Е.Л. Воспалительные заболевания мышц. Рациональная фармакотерапия ревматических заболеваний. 195–202. Москва. Литера. 2003.
2. Wortmann R.L. “Diseases of Skeletal Muscle” 1 с 45–57.
3. Dalakas M. Mechanisms of disease:signaling pathways and immunobyology of inflammatory myopathies. Nature clinical practice rheumatology. 2006, vol l2, 4, 219–227.
4. Насонов Е.Л., Штутман В.З, Саложин К.В, Гусева Н.Г., Насонова В.А, Плотц П. Клинико – иммунологическая гетерогенность идиопатических воспалительных миопатий. Клиническая Медицина 1995; 2 3–8.
5. Miller M. Clinical manifestations and diagnosis of adult dermatomyositis and po;ymyositis. UpToDate 2004; 12.2
6. Stahl, NI, Klippel, JH, Decker, JL. A cutaneous lesion associated with myositis. Ann Intern Med 1979; 91:577
7. Kovacs, SO, Kovacs, SC. Dermatomyositis. J Am Acad Dermatol 1998; 39:899.
8. Бондаренко И.В., Мухин Н.А., Насонов Е.Л. Поражение легких при полимиозите и дерматомиозите. Интерстициальные заболевания легких. Руководство для врачей. Под редакцией Ильковича М.М., Кокосова А.Н. 274–287. Санкт–Петерург. Нордмедиздат 2005.
9. Lakhanpal, S, Lie, JT, Conn, DL, Martin, WJ II. Pulmonary disease in polymyositis/dermatomyositis: A clinicopathologic analysis of 65 autopsied cases. Ann Rheum Dis 1987; 46:23.
10. Хитров А.Н. Поражение сердца при дерматомиозите и полимиозите. Автореферат диссертации на соискание ученой степени кандидата медицинских наук. Москва 1999.
11. Love, LA, Leff, RL, Fraser, DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: Myositisspecific autoantibodies define useful homogeneous patient groups. Medicine 1991; 70:360.
12. Bohan, A, Peter, JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med 1975; 292:403.
13. Bohan, A, Peter, JB, Bowman, RL, Pearson, CM. Computer–assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore) 1977; 56:255.
14. Reichlin, M, Arnett, FC. Multiplicity of antibodies in myositis sera. Arthritis Rheum 1984; 27:1150.
15. Love, LA, Leff, RL, Fraser, DD, et al. A new approach to the classification of idiopathic inflammatory myopathy: Myositisspecific autoantibodies define useful homogeneous patient groups. Medicine 1991; 70:360.
16. Plotz, PH, Rider, LG, Targoff, IN, et al. Myositis: Immunologic contributions to understanding cause, pathogenesis, and therapy. Ann Intern Med 1995; 122:715.
17. Brouwer, R, Hengstman, GJ, Vree Egberts, W, et al. Autoantibody profiles in the sera of European patients with myositis. Ann. Rheum Dis 2001; 60:116.
18. Mozaffar T, Pestronk, A. Myopathy with anti–Jo–1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry 2000; 68:472.
19. Гехт Б.М., Ильина Н.А. «Нервно–мышечные болезни». «Медицина» 1982.
20. Антелава О.А., Касаткина Л.Ф., Гуркина Г.Т., Хитров А.Н., Пикуля Н.В., Штутман В.З., Насонов Е.Л.. Дифференциальная диагностика мышечной слабости. Русский медицинский журнал N 14, 2004, 854–862.
21. Dalakas M.C. Polymyositis, dermatomyositis and inclusion–body myositis. Engl.J. Med. 1991, 325, 1487–1498.
22. Miller FW, Rider LG, Chung YL, et al, for the International Myositis Outcome Assessment Collaborative Study Group, Proposed preliminary core set measures for disease outcome assessment in adult and juvenile idiopathic inflammatory myopathies, Rheumatology (Oxford), 2001, 40,11,1262–1273.).
23. Griggs RC, Askanas, V, DiMauro, S, et al. Inclusion body myositis and myopathies. Ann Neurol 1995, 38:705.
24. Tanimoto K., Nakano K., Kano S. et al. Classification criteria for polymyositis and dermatomyositis. J. Rheumatol 1995; 22: 668–574.
25. Targoff I.N., Miller F.W., Medsger T.A. et al. Classification criteria for the idiopathic inflammatory myopathies. Curr Opin Rheumatol. 1997;9, 527–535.
26. Mastaglia FL, Phillips BA. Idiopathic inflammatory myopathies: epidemiology, classification, and diagnostic criteria.Rheum Dis Clin North Am 2002;28, 823–832.
27. Dalakas M.C., Hohlfeld R. Pofymyositis and dermatomyositis. Lancet 2003, 362, 971–982.
28. Nirmalananthan N, Holton JL. Hanna MG. Is it really myositis? Consideration of the differential diagnosis. Curr Opin Rheumatol 2004; 16: 684–691.
29. Ильина Н.И. «Миопатические синдромы» Клиническая медицина , 1983, N9, с 30–35.
30. Isenberg D.A., Allen E., Farewell V., et al. International consensus outcome measures for patients with idiopathic inflammatory myopathies. Development and initial validation of myositis activity and damage indices in patients with adult onset disease. Rheumatology (Oxford), 2004, 43, 1, 49–54.
31. Miller F.W., Rider L.G., Feldman. Development of validated disease activity and damage indices for the juvenile idiopathic inflammatory myopathies: I. Arthr. Rheum., 1997, 40, 11, 1976–1983.
32. Oddis C.V., Outcomes and disease activity measures for assessing treatments in the idiopathic inflammatory myopathies. Curr. Rheumatol. Rep. 2005, 7, 87–93.
33. Lovell D.J., Lindsley C.B., Rennebohm R.M., et al. Development of validated disease activity and damage indices for the juvenile idiopathic inflammatory myopathies. II. The Childhood Myositis Assessment Scale (CMAS): a quantitative tool for the evaluation of muscle function. The Juvenile Dermatomyositis Disease Activity Collaborative Study Group. Arthritis Rheum. 1999 42, 10, 2213–2219.
34. Park J. H., Olsen N. J, King L. et al. Use of imaging and P–31 magnetic resonance spectroscopy to detect and quantify muscle dysfunction in the myopathic variants of dermatomyositis. Arth.Rheum., 1995, 38, 68–77.
35. Lovell D.J., Lindsley C.B., Rennebohm R.M., et al. Development of validated disease activity and damage indices for the juvenile idiopathic inflammatory myopathies. II. The Childhood Myositis Assessment Scale (CMAS): a quantitative tool for the evaluation of muscle function. The Juvenile Dermatomyositis Disease Activity Collaborative Study Group. Arthritis Rheum. 1999 42, 10, 2213–2219.
36. Drake LA, Dinehart SM, Farmer ER, et al. Guidelines of care for dermatomyositis. J Am Acad Dermatol 1996; 34:824.
37. Fafalak RG, Peterson MGE, Kagen LJ. Strength in polymyositis and dermatomyositis: Best outcome in patients treated early. J Rheumatol. 1994; 21:643.
38. Joffe MM, Love LA, Leff RL et al. Drug therapy of the idiopathic inflammatory myopathies: Predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med 1993; 94:379.
39. Насонов Е.Л., Штутман В.З. Фармакотерапия идиопатических воспалительных миопатий. Клиническая фармакология, 1995, 4 (2). 57–63.
40. Grau JM, Herrero, C, Casademont J, et al. Cyclosporin A as a first choice therapy for dermatomyositis. J Rheumatol 1994; 21:381.
41. Qushmaq KA, Chalmers A, Esdaile JM. Cyclosporin A in the treatment of refractory adult polymyositis/dermatomyositis: population based experience in 6 patients and literature review. J Rheumatol 2000; 27:2855.
42. Антелава О.А., Соловьев С.К,. Хитров А.Н, Насонов Е.Л. Новые аспекты фармакотерапии идиопатических воспалительных миопатий. (Обзор литературы). Русский медицинский журнал 2006, 14, 8 (260), 627–629.
43. Антелава О.А., Насонов Е.Л. Место микофенолат мофетила при идиопатических воспалительных миопатиях. Научно–практическая ревматология 2006,3,38–41.
44. Dalakas, MC, Illa I, Dambrosia, JM, et al. A controlled trial of high–dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med 1993; 329:1993
45. Efthimiou P., Schwartzman S, Kagen L. Possible role for TNF–Inhibitors in the treatment of resistant dermatomyositis and polymiositis. Ann.Rheum. Dis. 2006, 13.
46. Hengstelman G.J., van den Hoogen F.H., Barrera P. et аl. Successful treatment of dermatomyositis and polymyositis with anti–tumor–necrosis–factor–alpha: preliminary observations. Eur.Neurol., 2003, 50(1),10–15.
47. Saaden C.K. Etanercept is effective in the treatment of polymyositis/dermatomyositis wich refractory to conventional therapy including steroid and other disease modifying agents. Arthr.Rheum., 2000, 43,193.
48. Hengstelman G., van den Hoogen F., van Engelen B., et al. Anti–TNF–blockade with infliximab (Remicaid) in polymyositis and dermatomyositis. Arthr.Rheum., 2000,43, 3193.
49. Levin T.D. Rituximab in the treatment of dermatomyositis. Arthr. Rheum. 2005, 52, 2, 601–607.
50. Levin T.D. A pilot study of rituximab therapy for refractory dermatomyositis. Arthr. Rheum. 2002,46, (suppl 9), 488.
www.rmj.ru
Воспалительная миопатия — Информационный медицинский портал о здоровье HealthInform г. Екатеринбург
Воспалительная миопатия — воспалительный процесс, возникающий преимущественно в скелетных мышцах и приводящий к их дегенеративным изменениям. Воспалительная миопатия характеризуется мышечной слабостью, болями в мышцах, резким снижением объема активных движений, развитием контрактур, уплотнением и отечностью мышц. Из методов диагностики воспалительной миопатии наиболее информативно определение уровня КФК и миоглобина, электромиография и биопсия мышц. Лечение воспалительной миопатии осуществляется высокими дозами глюкокортикостероидов с последующей поддерживающей терапией. Однако достаточно часто встречаются резистентные к кортикостероидам формы заболевания.
Воспалительные миопатии представляют собой целую группу заболеваний, в которую входят как системные поражение мышечной ткани, так и локальные воспалительные процессы в отдельных мышцах. Воспалительная миопатия системного характера представлена дерматомиозитом, полимиозитом, эозинофильным миозитом, миозитом с включениями, ревматической полимиалгией и миопатиями при системных заболеваниях. Примером локальной воспалительной миопатии может быть миозит глазных мышц, псевдотромбофлебит мышц голени и др. Кроме того, воспалительная миопатия может наблюдаться при некоторых инфекционных заболеваниях.
Возраст пациентов, наиболее подверженных заболеваемости, различается в зависимости от вида воспалительной миопатии. Так, полимиозит обычно возникает у лиц старше 30 лет. Дерматомиозит наблюдается как у взрослых, так и в детском возрасте. А миозит с включениями чаще встречается после 40 лет и является наиболее распространенной формой воспалительной миопатии в пожилом возрасте.
Причины возникновения воспалительной миопатии
В некоторых случаях воспалительная миопатия напрямую связана с инфекционным процессом. Агентами, вызывающими инфекционно-воспалительно поражение мышц, могут быть вирусы (краснуха, грипп, ВИЧ-инфекция, энтеровирусы и пр.), бактерии (чаще стрептококковые инфекции), паразитарные инвазии (цистицеркоз, токсоплазмоз, трихинеллез). При этом заболевание классифицируется как инфекционная воспалительная миопатия.
В тех случаях, когда прямая связь миопатии с инфекцией не прослеживается, говорят об идиопатической миопатии. К ней относятся: миозит с включениями, полимиозит, дерматомиозит, миопатии при системных заболеваниях (системной красной волчанке, склеродермии, синдроме Шегрена, системных васкулитах, ревматоидном артрите). Наиболее распространенным как в неврологии, так и в ревматологии является мнение, что идиопатическая воспалительная миопатия имеет аутоиммунный механизм развития. Однако до сих пор не выделен антиген, который запускает лежащую в ее основе аутоиммунную реакцию. В отдельных случаях воспалительная миопатия имеет семейный характер, свидетельствующий о ее генетическом происхождении.
Симптомы воспалительной миопатии
Воспалительная миопатия клинически проявляется болями в мышцах, прогрессирующей мышечной слабостью и тугоподвижностью в пораженных отделах конечностей. Болевой синдром (миалгия) возникает не только в период двигательной активности, но и при прощупывании мышц, а иногда и в состоянии полного покоя. Мышечная слабость, как правило, начинает проявляться затруднением при удерживании предметов в руках. По мере ее прогрессирования для пациента становится невозможным поднять руку или ногу, сесть, встать. Воспалительная миопатия распространенного характера часто приводит к значительному сокращению объема активных движений, которые может выполнять больной, а иногда и к полной обездвиженности.
Воспалительная миопатия протекает с отеком и уплотнением пораженных мышц. В результате воспаления в них происходят атрофические изменения, развивается миофиброз — замещение мышечных волокон на соединительнотканные, возможен кальциноз. Образующиеся мышечные контрактуры приводят к тугоподвижности суставов. Зачастую воспалительная миопатия имеет длительное ремиттирующее течение. В некоторых случаях, чаще у детей, наблюдается полное выздоровление после перенесенного острого приступа воспалительной миопатии.
Наиболее тяжело воспалительная миопатия протекает при вовлечении в воспалительный процесс мышц гортани и дыхательной мускулатуры, что ведет к развитию миопатического пареза гортани и дыхательной недостаточности. Слабость дыхательных мышц приводит к снижению легочной вентиляции и возникновению застойной пневмонии. Слабость мышц глотки является причиной дисфагии (расстройств глотания), при которой возможно попадание пищи в дыхательные пути и возникновение аспирационной пневмонии. Если воспалительная миопатия распространяется на глазодвигательные мышцы, то возникает косоглазие и опущение верхнего века. Поражение мимических мышц приводит к маскообразному выражению лица. Системная воспалительная миопатия, например, дерматомиозит и полимиозит, может сопровождаться поражением сердечной мышцы с клиникой миокардита, кардиомиопатии и сердечной недостаточности.
Полимиозит проявляется типичным для воспалительной миопатии мышечным синдромом, который захватывает мышцы проксимальных отделов конечностей, в первую очередь плечевого и тазового пояса. В большинстве случаев наблюдается также поражение внутренних органов: ЖКТ (диарея, запор, кишечная непроходимость, язва желудка с перфорацией или желудочно-кишечным кровотечением), сердца и сосудов (артериальная гипотония, аритмия, кардиомиопатия и др), легких (очаговая или долевая пневмония). В 15% случаев эта воспалительная миопатия сопровождается суставным синдромом в виде артритов суставов кисти, реже — других суставов конечностей.
Дерматомиозит имеет сходную с полимиозитом клиническую картину. Отличительной чертой является наличие кожных проявлений: эрите
healthinform.ru
Воспалительная миопатия
Воспаление скелетных мышц, приводящее к их дегенерации. Недуг проявляется мышечной слабостью, болями, резким снижением количества движений, ограничением пассивных движений в суставе, а также уплотнением и отечностью мышц. Воспалительные миопатии – целая группа системных и локальных заболеваний. Большинство типов этого заболевания проявляется после тридцати лет, правда образование дерматомиозита возможно и у детей.
Причины воспалительной миопатии
Мотивы развития болезни связаны с типом недуга. Воспалительную инфекционную миопатию способны спровоцировать вирусы (краснухи, гриппа, ВИЧ-инфекции, энтеровирусов и др.), бактерии (в основном, стрептококки), паразитарные инвазии (цистицеркозы, токсоплазмозы, трихинеллезы).
Если образование миопатии никак не коррелирует с инфекционными возбудителями, то ее называют идиопатической. Эта группа заболеваний может выражаться миозитом с включениями, полимиозитом, дерматомиозитом, а также миопатиями сопровождающими системные заболевания (волчанка, склеродермия, синдром Шегрена, системный васкулит, ревматоидный артрит). Считается, что заболевание может передаваться по наследству или развиваться на фоне аутоиммунного процесса. Правда, что касается последнего, то антиген, запускающий аутоиммунный процесс пока не найден.
Симптомы воспалительной миопатии
Признаками недуга считаются: мышечные боли, нарастающая слабость в мышцах, тугоподвижность воспаленных отделов рук или ног. Болевой синдром наблюдается в момент движений, прощупывания и в период покоя. Мышечная слабость выражается сложностью в удержании предмета в руках, но после нарастания симптоматики, больной и вовсе не в состоянии поднимать конечности, вставать или садиться. В редких случаях недуг полностью обездвиживает человека, чаще – сокращает объем движений, которые он способен выполнять.
Пораженные заболеванием мышцы отекают и уплотняются. По мере прогрессирования миопатии они атрофируются. Воспаление может привести к замещению мышечной ткани соединительной или к отложениям солей кальция в волокнах мышц. Такие патологии, в свою очередь провоцируют развитие тугоподвижности суставов.
Зависимо от типа недуга и локализации воспаленных мышц, развиваются специфические симптомы.
Диагностика воспалительной миопатии
Как правило, за консультацией обращаются к неврологу, но он, зависимо от типа недуга, может подключать ревматолога, кардиолога, пульмонолога, дерматолога, иммунолога и докторов других направлений. Как правило, больной сдает биохимический анализ крови, правда его показатели не всегда взаимосвязаны со степенью поражения мышц. Имея лишь данные полученные из анализа крови, врачам сложно судить о прогрессировании заболевания. Потому они исследуют мышечную силу в динамике, проводят ЭМГ и биопсию пораженных мышц. Ввиду того, что воспалительной миопатии часто сопутствуют патологии внутренних органов специалисты могут назначать рентгенографию легких, электрокардиографию, эхокардиографию, гастроскопию, ультразвуковое исследование.
Лечение воспалительной миопатии
Общеустановленной терапией недуга является лечение стероидными гормонами, которые синтезирует кора надпочечников – глюкокортикоидными препаратами. Обычно, применятся Преднизолон, дневная дозировка которого от 80 до 100 миллиграмм. После улучшения состояния пациента, которое проявляется увеличением силы в мышцах, дозу снижают до 15 мг в сутки. Если болезнь протекает тяжело, больному внутривенно вводят высокие дозы Метилпреднизолона. Длительную терапию осложняют побочные эффекты гоюкокортикостероидов, а также устойчивость к гормональными медикаментам.
Если через 90 дней приема гормональных препаратов мышечная сила не увеличилась, применяют альтернативные методы терапии. К примеру, пациенту вместо стероидных гормонов или в дополнение к ним назначается прием Азатиоприна, Метотрексата, Циклоспорина или Циклофосфамида.
При системной воспалительный миопатии аутоиммунного характера больному проводят плазмоцитофорез – процедуру забора крови, ее очищения и возврата обратно в кровоток.
Профилактика воспалительной миопатии
Профилактические меры включают в себя предотвращение заражения ВИЧ-инфекцией, краснухой, энтеровирусом, а также стрептококками и паразитарными болезнями, которые могут вызвать образование миопатии.
www.obozrevatel.com