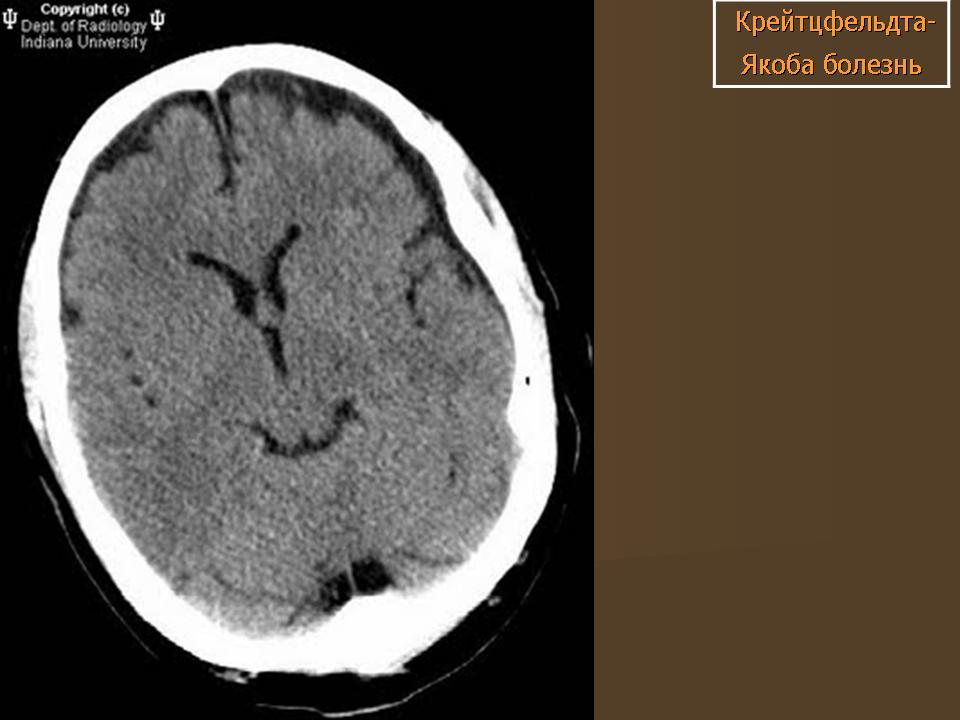
Болезни крейтцфельдта якоба: новый взгляд на старую проблему (клиника, диагностика, прогноз, лечение)
причины, симптомы и лечение в статье невролога Толмачев А. Ю.
Над статьей доктора Толмачева Алексея Юрьевича работали литературный редактор Юлия Липовская, научный редактор Сергей Федосов и шеф-редактор Лада Родчанина
Дата публикации 22 октября 2019Обновлено 8 сентября 2022
Определение болезни. Причины заболевания
Болезнь Крейтцфельдта – Якоба — редкое и смертельное дистрофическое заболевание коры и подкорковых центров головного и спинного мозга. Вызывается болезнетворным белком прионом с аномальной третичной структурой (с особым не свойственным для человека вариантом последовательности аминокислот, из которых состоит белок). Возникает под воздействием внешних и наследственных факторов, не исследованных в полной мере.
Заболевание приводит к развитию атрофии мозга и формированию губчатой энцефалопатии (коровьего бешенства), проявляется прогрессирующим снижением интеллекта, двигательной активности и другими многочисленными неврологическими симптомами, не являющимися строго специфичными только для этого заболевания. Поэтому по симптомам поставить этот диагноз нельзя [1][2].
Термин болезнь Крейтцфельдта – Якоба был введен в 1922 году после докладов немецких врачей Ханса Герхарда Крейсфельдта (1920 год) и Альфонса Марии Якоба (1921 год). В докладах сообщалось о шести случаях нового нейродегенеративного заболевания [8].
Причиной всех форм заболевания является белок прион, который не имеет в своем составе ДНК или РНК, поэтому не является микроорганизмом и классическим инфекционным агентом (вирусом, бактерией, простейшим или грибком). Прион — это протеин (сокращенно — PrP), который в норме входит в состав нервных клеток, и его функция до конца не известна. Заболевание начинается при появлении в организме измененной формы данного протеина (сокращенно — PrPres), который из-за своего особого пространственного строения становится невосприимчивым к ферментам и может сворачиваться с формированием «палочек». В таком виде он способствует разрушению клетки и накапливается в организме, вызывая каскад болезненных реакций. При этом иммунная система его игнорирует [1].
Заболевание начинается при появлении в организме измененной формы данного протеина (сокращенно — PrPres), который из-за своего особого пространственного строения становится невосприимчивым к ферментам и может сворачиваться с формированием «палочек». В таком виде он способствует разрушению клетки и накапливается в организме, вызывая каскад болезненных реакций. При этом иммунная система его игнорирует [1].
Среди причин появления в организме болезнетворного прионного белка выделяют:
- Спонтанную трансформацию собственного белка PrP человека в патологический PrPres у людей с генетической предрасположенностью под влиянием не полностью исследованных инициирующих факторов.
- Попадание болезнетворного инфекта экзогенно (из внешней среды):
- через пищеварительный тракт при поедании пищи животного происхождения — как свежих, так и консервированных. Особенно велико содержание инфекционного агента в мясе, мозге, крови и селезёнке забитых коров.
 Кроме коровьего мяса возможны экзотичные случаи заражения при ритуальном каннибализме;
Кроме коровьего мяса возможны экзотичные случаи заражения при ритуальном каннибализме; - при трансплантации органов: сердца, печени, почек, костного мозга, роговицы глаза, при переливании цельной крови или её компонентов: плазмы, эритроцитарной и лейкоцитарной массы;
- при использование лекарственных препаратов из животной ткани, в том числе сыворотки крови крупного рогатого скота (коров), либо же изготовленных на основе гипофиза животных;
- возможна передача заболевания медицинскому персоналу и родственникам больного через биологические жидкости больного человека (кровь и плазму) при условии нарушения целостности кожных покровов или попадании инфекционного белка в желудочно-кишечный тракт. Поэтому при уходе за больным необходимо соблюдение санитарно-эпидемиологических правил. Установлено, что заболевание не передается воздушно-капельным путем и через прикосновения. Возможна передача через мокроту, кал, мочу, однако исследований об этом мало.
- Наследственная передача заболевания.
 Кроме коровьего мяса возможны экзотичные случаи заражения при ритуальном каннибализме;
Кроме коровьего мяса возможны экзотичные случаи заражения при ритуальном каннибализме;
Наиболее часто заболевание проявляется в возрасте 60-65 лет. Отличается быстрым течением, большинство больных умирает в течении первого года заболевания. В редких случаях возникают формы с быстрым (несколько недель) развитием акинетического мутизма — отсутствия речи и двигательной активности. В данном случае смерть чаще наступает в течении первых двух месяцев болезни. Бывают формы с медленным течением (около двух и более лет) [1][4].
Случаев выздоровления у пациентов с диагносцированной болезнью Крейтцфельдта – Якоба не зарегистрировано, смертность составляет 100%.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением — это опасно для вашего здоровья!
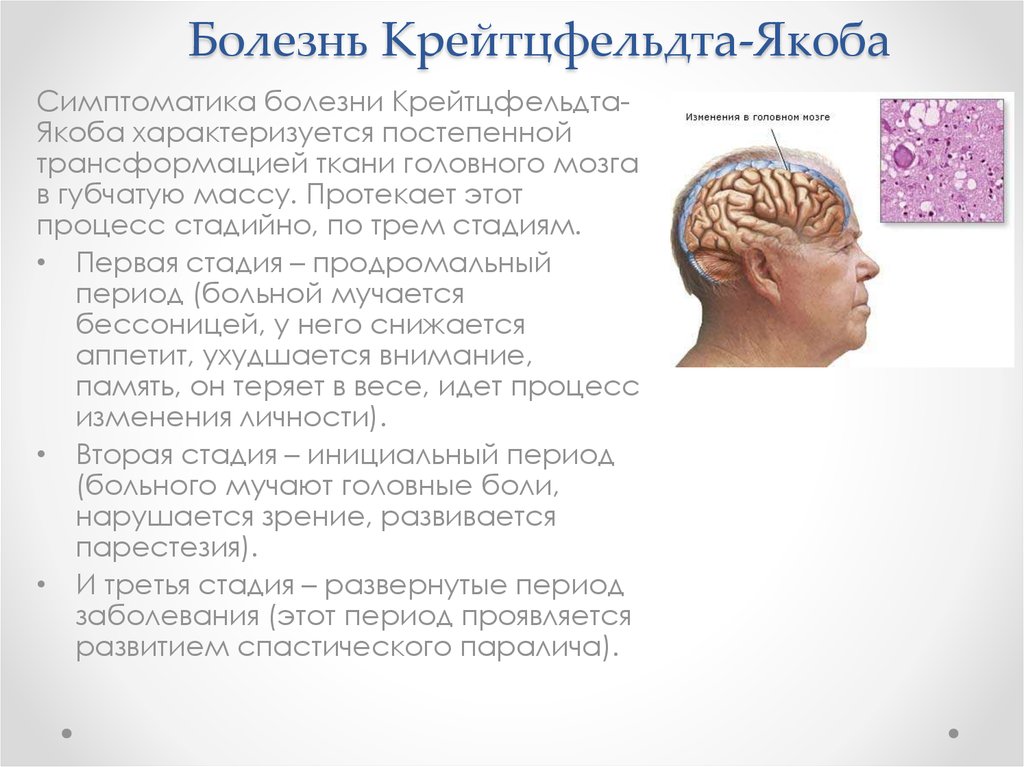
Симптомы болезни Крейтцфельдта — Якоба
Выделяют три стадии болезни Крейтцфельдта – Якоба:
- продромальная;
- развернутых клинических проявлений;
- терминальная (финальная) стадия.

В продромальной стадии отсутствует какая либо специфическая клиническая картина. Выделяют вегетативные и астенические нарушения: слабость, быструю утомляемость, проблемы со сном и нарушение аппетита, снижение веса, сексуальную дисфункцию и болевые ощущения без чёткой локализации. Могут отмечаться лёгкие нарушения высшей нервной деятельности: снижение памяти и внимания, ухудшение мышления. Однако эти изменения чаще расцениваются как проявления астении. Возможно появление психических нарушений: безучастности, апатии, бредовых идей (вплоть до психоза), стартл-реакций — непроизвольных реакций испуга и вздрагивания на внешние раздражители.
Чаще всего начальными проявлениями заболевания являются различные нарушения зрения: отсутствие чёткости и двоение в глазах, сужение полей зрения, нарушение узнавания зрительных образов (лиц, предметов), затруднение пространственной ориентации.
По некоторым данным 35 % случаев начинается с депрессивных нарушений и проблем с памятью, в 34 % страдает координация движений и зрение, в 21 % отмечается сочетание симптомов [1].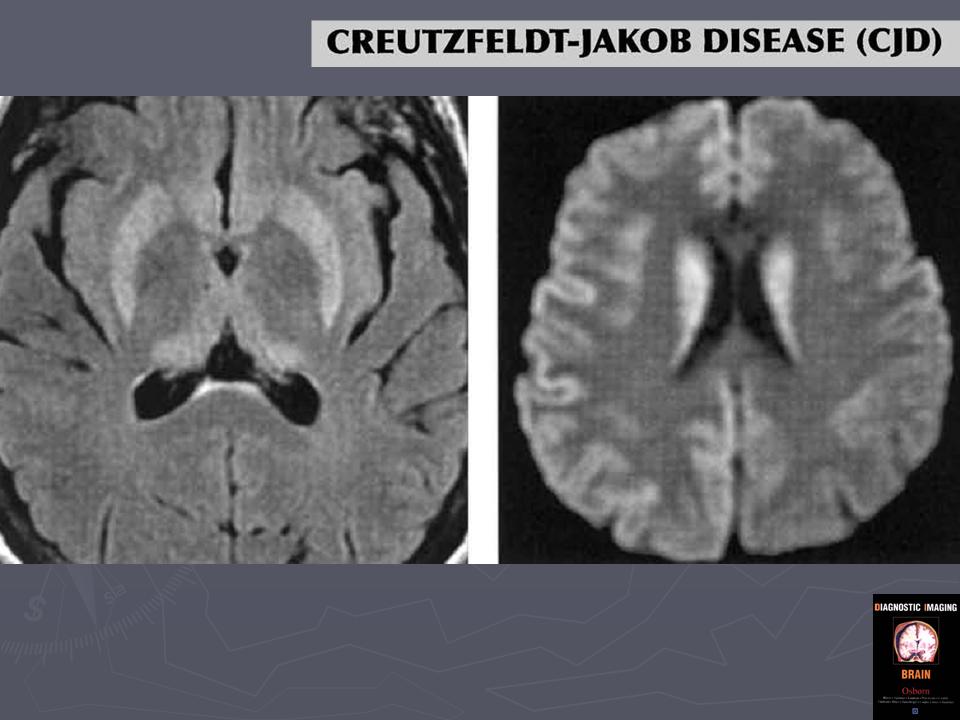
К основным симптома в развернутой стадии болезни Крейтцфельдта – Якоба относят: быстро прогрессирующее слабоумие (деменцию) и подёргивания одиночных или нескольких мышц конечностей и туловища. Эпилептические припадки встречаются редко.
На финальной стадии заболевания отмечается выраженное слабоумие. Больной не разговаривает и не способен к целенаправленным движениям, наблюдаются параличи или некоординированные насильственные движения. Также движения затруднены из-за значительного повышения мышечного тонуса, до степени ригидности. Распространены мышечные сокращения, прогрессируют нарушения со стороны сердечно-сосудистой и дыхательной системы. Чаще всего смерть наступает от дыхательной недостаточности [1][2][3][4].
Патогенез болезни Крейтцфельдта — Якоба
Выделяют два этапа патогенеза болезни Крейтцфельдта – Якоба: экстрацеребральный (не относящийся к мозгу, находящийся за его пределами) и церебральный [1]. При этом недостаточно хорошо исследован патогенез наследственной и спорадической формы болезни.
При этом недостаточно хорошо исследован патогенез наследственной и спорадической формы болезни.
Экстрацеребральный этап патогенеза. Болезнетворный прионный белок попадает в организм и не поступает сразу в нервную ткань, а попадает в органы лимфатической системы, используя в качестве переносчиков клетки, обеспечивающие иммунные реакции организма: лимфоциты, макрофаги и дендритные клетки. В лимфатических узлах и селезёнке белок самовоспроизводится, преобразуя нормальные для организма человека белковые молекулы в себе подобные. Естественного иммунного ответа на патологический агент не происходит, так как прионный белок, обладая структурным сходством с физиологичным для человека, не воспринимается как чужеродный. Сам прион не оказывает какого-либо повреждающего действия на лимфатические узлы, миндалины и селезёнку, не вызывает их увеличения, воспалительных или других специфических структурных изменений.
Считается, что за дальнейший перенос болезнетворного белка в спинной и головной мозг ответственна вегетативная нервная система, так как её симпатические волокна в основном и иннервируют (пронизывают нервными окончаниями) лимфоидные органы.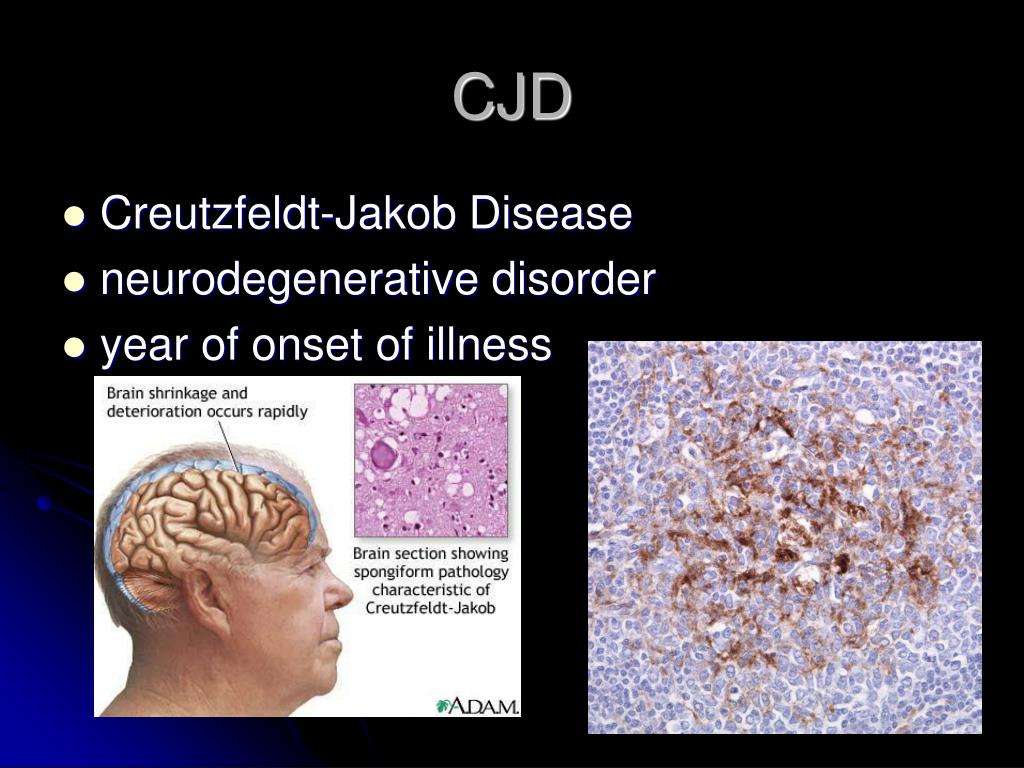 Также допускается проникновение прионного белка через гематоэнцефалический барьер, минуя лимфатическую систему. При передаче прионного белка при нейрохирургических или офтальмологичесих операциях заражение происходит минуя желудочно-кишечный тракт: инфекционный агент попадает непосредственно в нервную ткань.
Также допускается проникновение прионного белка через гематоэнцефалический барьер, минуя лимфатическую систему. При передаче прионного белка при нейрохирургических или офтальмологичесих операциях заражение происходит минуя желудочно-кишечный тракт: инфекционный агент попадает непосредственно в нервную ткань.
Церебральный этап патогенеза. После попадания в нервные клетки прионные молекулы приобретают патогенные свойства. За счёт особого пространственного строения болезнетворные белки не включены в естественный метаболизм клетки и поэтому не разрушаются ферментами, а накапливаются в жидком содержимом клетки (цитозоле) и нервных окончаниях (синапсах). Из-за присутствие патогенного белка уменьшается содержание физиологичного прионного белка (PrP), который обеспечивает нормальные процессы питания нервной клетки.
В результате патологического процесса прогрессирует атрофия и утрата нервных клеток, скопление прионного белка отмечается не только в гибнущих клетках, но и в межклеточном пространстве.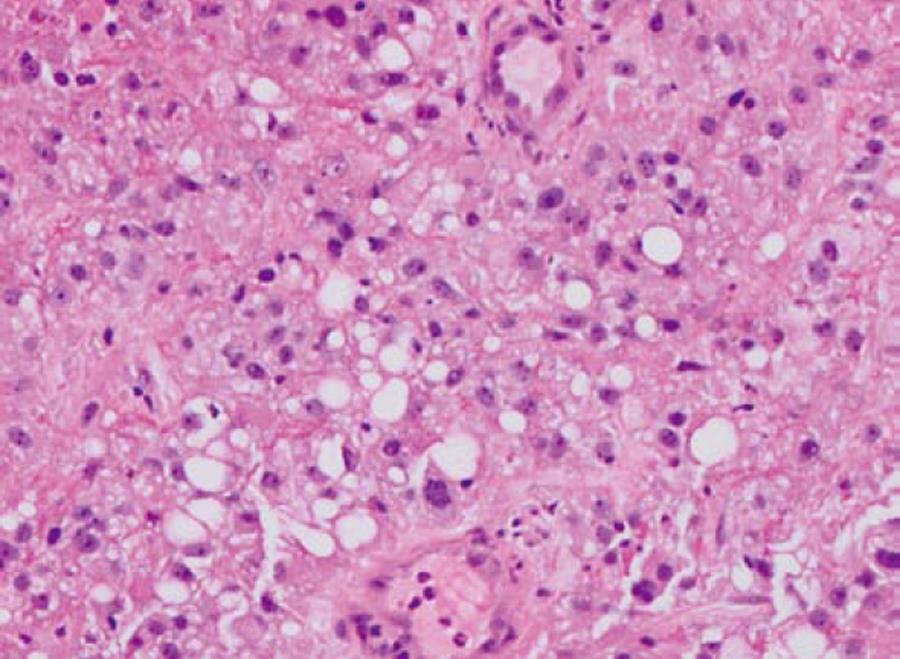 Изменяется структура мозга за счёт очагов дегенерации, происходит его губчатая трансформация, формируется спонгиформная (губчатая) энцефалопатия [5].
Изменяется структура мозга за счёт очагов дегенерации, происходит его губчатая трансформация, формируется спонгиформная (губчатая) энцефалопатия [5].
Классификация и стадии развития болезни Крейтцфельдта — Якоба
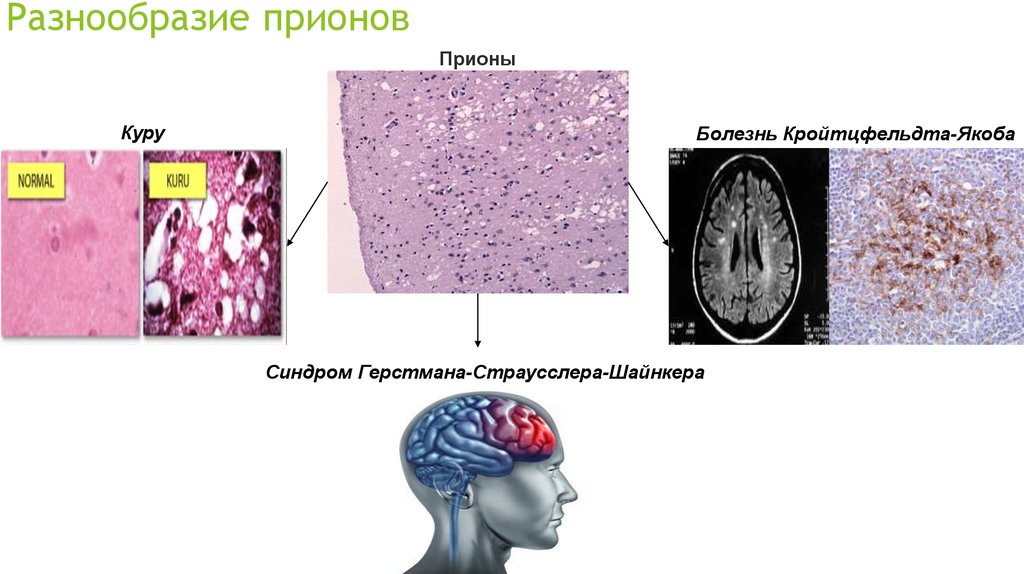
В первое время после открытия из-за разнообразия симптомов и вариантов течения врачи сомневались, что болезнь Крейтцфельдта – Якоба — это самостоятельное заболевание [8]. Сейчас в зависимости от возникновения белка в организме выделяют четыре формы болезни (куру так же причисляется некоторыми авторами к форме болезни Крейтсфельдта – Якоба):
- Спорадический (спонтанный, классический) — соматическая мутация PRNP или спонтанная конверсия РгР в PrPSс без видимой внешней причины. Частая встречаемость данной формы возможно связана с сложностями сбора анамнеза и установления ретроспективной связи с другими причинами [8];
- Приобретенный (инфекционный ятрогенный) — заражение путем инокуляции (прививки), то есть непосредственное заражение человека через введение болезнетворного приона при переливании цельной крови или ее компонентов, трансплантации органов и тканей, использование зараженного медицинского инструментария во время оперативных вмешательств или медицинских манипуляций [9];
- Приобретенный (инфекционный) — новый вариант заболевания, болезнь бешенства коров, также её называют «вариантным подтипом». Предполагает алиментарный путь заражение при употреблении мяса больных животных;
- Приобретенный (инфекционный, куру) — алиментарный путь заражения, в основном в племенах Папуа Новой-Гвинеи, где был распространен ритуальный каннибализм. В связи с запретом ритуального каннибализма сейчас практически не встречается [9];
- Наследственный (семейный) — передаваемая наследственным путём мутация PRNP.
 Предполагает алиментарный путь заражение при употреблении мяса больных животных;
Предполагает алиментарный путь заражение при употреблении мяса больных животных;По преобладающему расположению спонгиформных очагов различают редкие подтипы, которые, по сути, являются вариантами спорадической формы [1][8]:
| № | Подтип заболевания | Преимущественная локализация очагов | Клиника |
|---|---|---|---|
| 1 | Миоклонический или диффузный церебральный | Относительно равномерное распределение возбудителя в центральной нервной системе | Преобладают миоклонические подергивания (резкие, в течении 1-2 секунд, внезапные, непроизвольные, повторяющиеся сокращения отдельных мышечных групп, по типу вздрагивания), и нарушения высшей нервной деятельности |
| 2 | Подтип Хейденхайна (Heidenhain) | В коре затылочных догей головного мозга | Слепота и нарушение узнавания зрительных образов, связанные с поражением корковых зон зрительного анализатора |
| 3 | Дискенетический подтип (гиперкенетический — связанный с развитием насильственных, повторяющихся движений) с мышечной атрофией или без | В подкорковых ганглиях, спинном мозге, корешках спинного мозга, периферических нервах | Развитие гиперкинезов (непроизвольных движений), мышечной дистонии, в некоторых случаях ранних параличей с атрофическими изменениями конечностей |
| 4 | Таламический подтип | Поражение таламических ядер | Нарушение сознания и внимания, психические нарушения, параличи, нарушения чувствительности |
| 5 | Подтип Броунелла-Оппенгейма (атактический — то есть, связанный с нарушением координационно двигательной функции) | С наибольшим поражением мозжечка | Выраженные головокружения, нарушение равновесия при сравнительно лёгком поражении функции высшей нервной деятельности: памяти, мышления, психики |
| 6 | Панэнцефалитический (японский) подтип | Обширное поражение с дегенерацией белого вещества, вакуолизацией (дистрофическим процессом, сопровождающимся возникновением в клетке пузырьков с жидкостью — вакуолей) и губчатой трансформацией серого вещества | Относительно более длительная форма течения. Характерны акинетический мутизм, стартл-реакции, эпиприпадки, стоны Характерны акинетический мутизм, стартл-реакции, эпиприпадки, стоны |
В развитии болезнь Крейтцфельдта – Якоба проходит пять стадий[1]:
- Продромальная стадия.
- Стадия первых симптомов.
- Развернутая стадия.
- Финальная стадия.
- Стадия продлённой жизни в условиях реанимационного отделения.
По течению различают три варианта развития заболевания:
- Относительно медленно развивающиеся в развернутой стадии заболевания.
- Нарушения интеллекта и поведения, которые в последующем быстро прогрессируют.
- Ступенеобразное или персистирующее, неуклонное прогрессирование заболевания без периодов стабилизации.
- Быстрое ухудшение симптоматики в течение нескольких месяцев с последующим относительно более медленным развитием заболевания в терминальной стадии.
Осложнения болезни Крейтцфельдта — Якоба
Осложнения определяются выраженностью и прогрессированием ведущих симптомов заболевания, которые возникают из-за нарушения центральной нервной регуляции внутренних органов:
- Эпилептический статус чаще по бессудорожному типу, развивающийся в развернутой стадии болезни [6]
- Острые психозы, требующие профильного лечения.
- Инфекционные осложнения (например, застойная пневмония), связанные с обездвиженностью больных.
- Трофические осложнения: атрофии, пролежни, развивающиеся как следствие денервации кожи и мышечной ткани.
- Сердечная и дыхательная недостаточность, возникающая чаще в финальной стадии заболевания.

Диагностика болезни Крейтцфельдта — Якоба
Ранняя диагностика заболевания затруднена. В основном предварительный диагноз ставится клинически или на основании полученных данных о уже выявленных семейных случаях. При подозрении на болезнь Крейтцфельдта – Якоба возможно проведение генетического анализа.
В целом постановка диагноза остается сложной проблемой, так у болезни отсутствуют какие-либо специфические изменения в картине общего и биохимического анализа крови. Иногда отмечается повышение печёночных трансаминаз (ферментов, которые дают наиболее корректные данные для лабораторной диагностики проблем печени) и щелочной фосфатазы (фермента для лабораторной диагностики заболеваний костей, печени и сердца), что тоже не специфично только для болезни Крейтцфельдта – Якоба.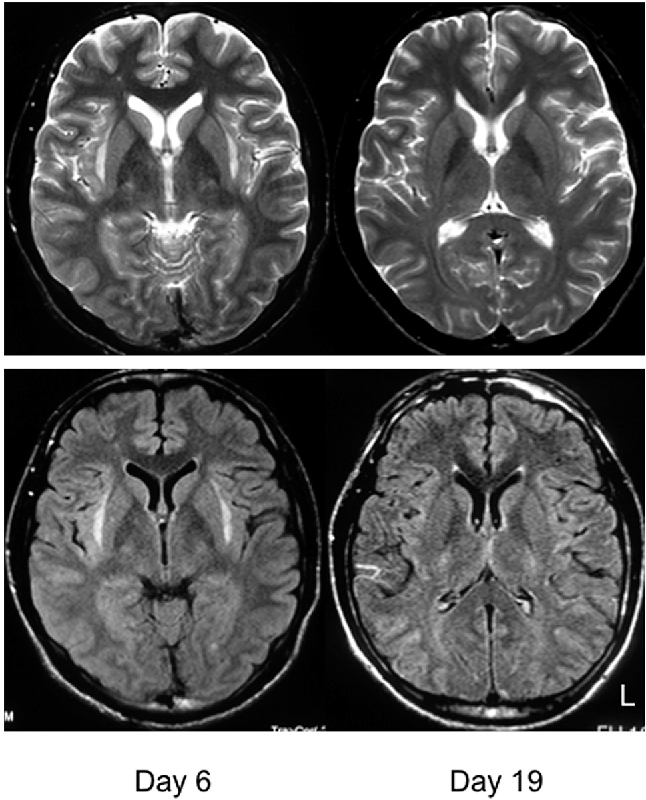 Отсутствуют и специфические иммунологические маркеры.
Отсутствуют и специфические иммунологические маркеры.
Окончательный диагноз ставится на основании биопсии или аутопсии. Ранее достоверным считалось исследование только тканей мозга, однако сейчас описывают варианты исследований биоптата нёбных миндалин. Также используют биологические методы — здоровая нервная ткань лабораторных животных заражается исследуемой тканью больного, с последующим выяснением наличия заражения у животного [1][5].
В комплексе с клиническими данными и историей болезни при постановке диагноза помогает электроэнцефалография. Уже на ранних этапах болезни замедляется биоэлектрическая активность мозга. По мере прогрессирования заболевания отмечается появление периодических двух или трёхфазных острых волн с частотой около 2Гц. Но эти изменения не характерны только для прионных болезней и могут встречаться при различных заболеваниях мозга, возникающих при нарушении обмена веществ. При некоторых генетических вариантах болезни Крейтцфельдта – Якоба изменения электроэнцефалограммы вовсе отсутствуют.
При некоторых генетических вариантах болезни Крейтцфельдта – Якоба изменения электроэнцефалограммы вовсе отсутствуют.
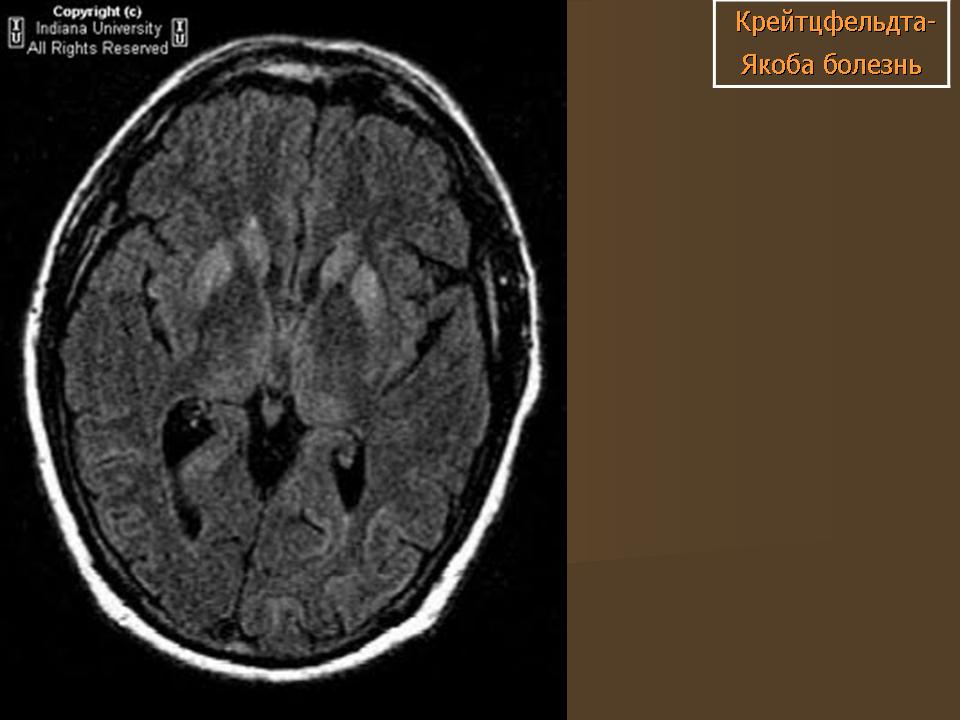
При провединии МРТ или позиционно эмиссионной томографии можно обнаружить дегенеративные изменения головного мозга. Данные изменения так же не являются определяющими и встречаются при других патологиях нервной системы [1][5].
Состав спинномозговой жидкости довольно длительное время при заболевании может оставаться нормальным. После выделяется специфичный белок, который проявляется именно при болезни Крейтцфельдта – Якоба, но он встречается и при нарушениях мозгового кровообращения и воспалительных заболеваниях нервной системы. Однако если других симптомов воспаления нет, можно предположить именно болезнь Крейтцфельдта – Якоба. На поздних стадиях в спинномозговой жидкости обнаруживается нейрональная енолаза — активное вещество, участвующее в обмене углеводов, которое встречается при распаде нервных клеток. Присутствие нейрональной енолазы также не является специфичной только для прионных болезней, её повышение отмечается при опухолях коры головного мозга и новообразованиях щитовидной железы.
Присутствие нейрональной енолазы также не является специфичной только для прионных болезней, её повышение отмечается при опухолях коры головного мозга и новообразованиях щитовидной железы.
Таким образом, несмотря на развитие генетической и иммунохимической диагностики, прижизненная диагностика болезни Крейтцфельдта – Якоба остаётся проблемой — из-за многообразия форм развития, клинических проявлений и вариантов течения.
Лечение болезни Крейтцфельдта — Якоба
Эффективное лечение причины прионных заболеваний не разработано [14]. Не существует вариантов лекарственного воздействия на этапы развития болезни Крейтцфельдта – Якоба, приводящих к значимому замедлению и стабилизации процесса. Известные иммунокорегирующие (противовирусные, гормональные, противоопухолевые) средства не показали значимого влияния на прогноз.
Небольшое влияние на динамику развития заболевания имеют следующие препараты:
- брефельдин А — разрушает аппарат Гольджи (структуру внутри клетки, которая отвечает за завершение синтеза и выведение органических веществ, продуцируемых в клетке), замедляя образование прионного белка в зараженной культуре клеток;
- кетамин, фенциклидин и другие блокаторы кальциевых каналов (особенно NMDA-рецепторов) способствуют большей выживаемости нервных клеток;
- тилорон — искусственно синтезированное вещество относящееся к группе иммунокорректоров, при длительном использовании способствует накоплению гликозаминогликанов в клетках, снижает скорость накопления прионных белков. Одновременно тилорон нарушает обмен фосфолипидов и ухудшает стабильность клеточных мембран, что может привести к нарушению клеточного обмена и гибели клетки.
 Одновременно тилорон нарушает обмен фосфолипидов и ухудшает стабильность клеточных мембран, что может привести к нарушению клеточного обмена и гибели клетки.
Одновременно тилорон нарушает обмен фосфолипидов и ухудшает стабильность клеточных мембран, что может привести к нарушению клеточного обмена и гибели клетки.Вышеуказанные препараты так и не нашли клинического подтверждения своей эффективности в связи с частотой возникновения побочных эффектов, несопоставимых с возможной пользой от длительного использования.
Ведутся исследования следующих направлений в лечении [12]:
- методы, способствующие медикаментозному обратному преобразованию болезнетворного прионного белка в нормальный человеческий;
- медикаменты, предотвращающие пространственную трансформацию здоровых белков человека в патологические, что останавливало бы дальнейшее развитие заболевания;
- фильтры для удаления патологического прионного белка из крови.
Симптоматическое лечение пациентов с болезнью Крейтцфельдта – Якоба направлено на повышение уровня жизни и облегчение симптомов заболевания:
- Антиконвульсанты — группа препаратов, используемых для предотвращения или уменьшения проявления эпилептических пароксизмов;
- Антидепрессанты и другие психокорректоры для лечения эмоциональных и поведенческих нарушений, корректоры сна;
- Антибактериальные препараты при развитии вторичных инфекционных заболеваний у обездвиженных пациентов;
- Лечебные мероприятия, направленные на продление жизни в условиях реанимационного отделения: оксигенотерапия, искусственная вентиляция лёгких, препараты для стимуляции дыхательной и сердечно-сосудистой системы.
Прогноз. Профилактика
Прогноз для жизни неблагоприятный. В зависимости от формы и варианта течения смерть от заболевания наступает в период от двух месяцев до полутора лет.
Профилактика болезни Крейтцфельдта – Якоба имеет ряд сложностей медикосоциального и юридического характера. Например, человек с наследственной формой болезни несмотря на высокий риск передачи заболевания может сохранять желание иметь собственных детей. Ухудшают ситуацию недостаточная разработка профилактических мероприятий из-за многообразия форм заболевания и отсутствия единого научного подхода к профилактике. Если носительство мутагенного гена выявлено, на данном этапе развития науки нет возможности уменьшить вероятность заболеть. Нет юридических механизмов, позволяющих отслеживать пациентов с возможной ранней стадией заболевания для профилактики возможной передачи заболевания от человека к человеку, например при его участии в донорстве крови или иных органов.
Государственная профилактика распространения приобретенных прионных заболеваний на территории России предполагает [15]:
- Поставку мяса в РФ только после официальных запросов об отсутствии в странах поставщиках спонгиформной энцефалопатии.
- Изучение санитарно эпидемической обстановки на территории России для своевременного выявления случаев спонгиформной энцефалопатии.
- Запрет регистрации, перерегистрации, а также исключение из «Государственного реестра лекарственных средств» препаратов, производимых из гипофиза человека. Их заменяют синтетическими аналогами.
- Взаимодействие с Всемирной Организацией Здравоохранения по вопросу мониторинга и предупреждения прионных заболеваний.

Профилактика непосредственной передачи прионных заболеваний «от человека к человеку»:
- Инструменты после проведения любых манипуляций подлежат уничтожению, если это невозможно, проводится длительная санитарная обработка.
- Лечебные, диагностические и патологоанатомические манипуляции с пациентами, у которых подозревается или установлено прионное заболевание, проводятся в кольчужных перчатках, масках и очках. Особенно это важно при аутопсии и любых манипуляциях с трупами, так как после смерти пациента прионный белок остается заразным много лет.
Постоянно идет разработка и поиск новых методов профилактики и предотвращения заражения прионными инфекциями, включая выведение новых мясных пород сельскохозяйственных животных, неспособных к передаче прионного белка [1][11].
Болезнь Крейтцфельдта–Якоба
Это быстро прогрессирующее фатальное нейродегенеративное заболевание, ключевым патогенетическим звеном которого является гибель нейронов, индуцированная прионовыми белками. Заболевание приводит к быстро прогрессирующему слабоумию и смерти обычно в течение года от начала. Подавляющее большинство носит спорадический характер, но изредка встречаются семейные и приобретенные формы.
ИсторияЗаболевание было названо в честь Ханса Герхарда Кройцфельдта (1885-1964), немецкого невролога, который впервые описал это состояние в 1920 году, и Альфонса Марии Якоба (1884-1931), немецкого невролога, которая также описала это состояние в отдельном исследовании в 1921 году.
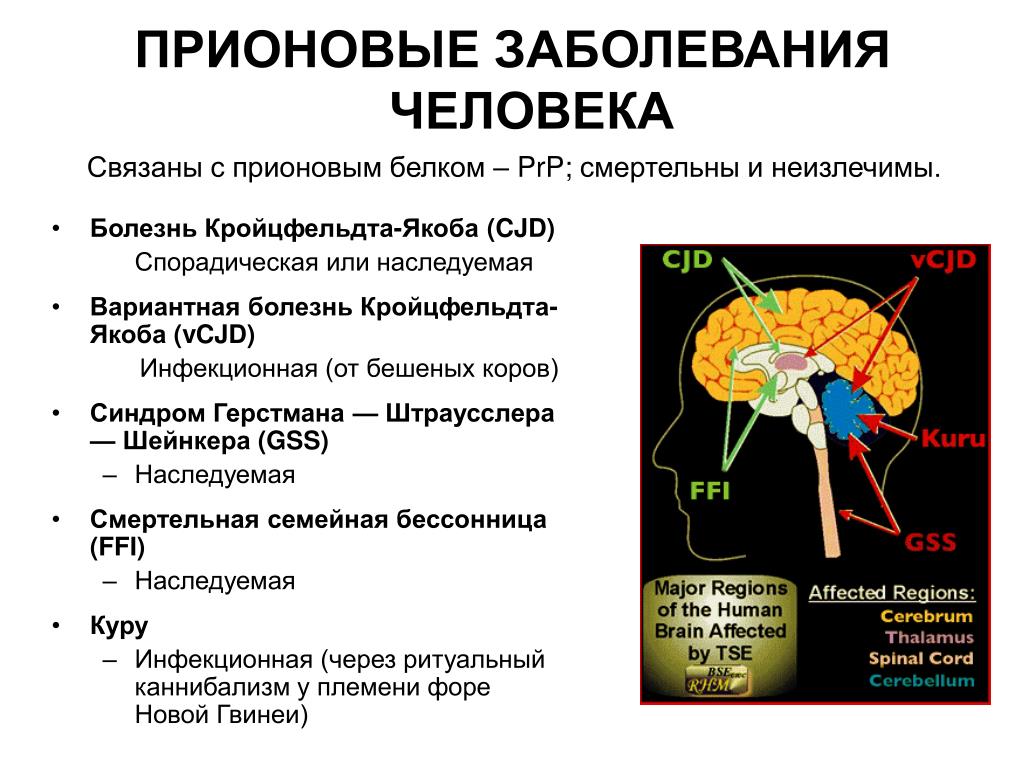
ЭпидемиологияОписаны три основных типа болезни Крейтцфельдта-Якоба:
- Спорадический – составляет 85-90% случаев и разделена на множество подтипов в зависимости от мутации;
- Семейный – 10% случаев, вызвана мутацией PRPc;
- Ятрогенный;
К основным клиническим проявлениям заболевания относятся: быстро прогрессирующие когнитивные нарушения, миоклонус, дистония, акинетикоригидный синдром, спастичность, гиперрефлексия, атаксия, зрительные расстройства, на поздних этапах заболевания акинетический мутизм.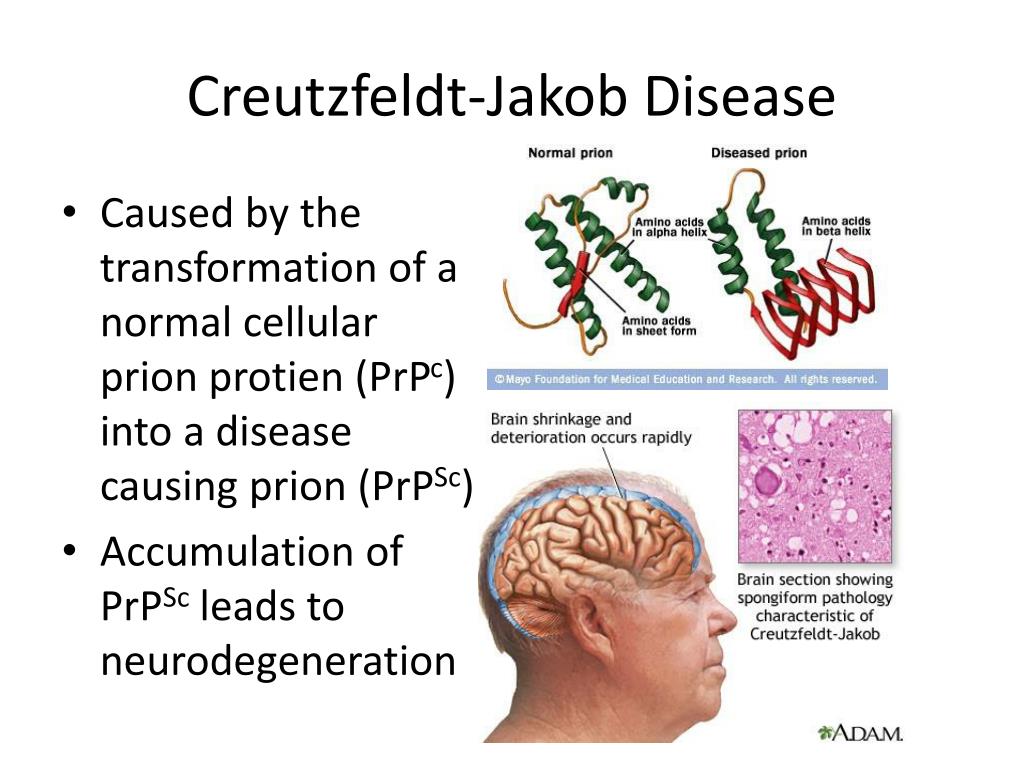 Примерно в трети случаев отмечаются эпилептические припадки. Средняя продолжительность жизни при спорадической форме БКЯ составляет около 5 мес; более 90% пациентов умирают в течение 1 года из-за аспирационной пневмонии в состоянии акинетического мутизма. Используемые в настоящее время диагностические критерии БКЯ включают быстро прогрессирующую деменцию, экстрапирамидно/ пирамидные и зрительные расстройства, миоклонус, мозжечковую атаксию, а также характерные изменения ЭЭГ в виде комплексов высокоамплитудных 2–3-фазных острых волн и положительный маркер белок 14-3-3 в цереброспинальной жидкости (ЦСЖ). Сложности диагностики БКЯ связывают с редкостью заболевания, клиническим полиморфизмом и недостаточной информированностью врачей.
Примерно в трети случаев отмечаются эпилептические припадки. Средняя продолжительность жизни при спорадической форме БКЯ составляет около 5 мес; более 90% пациентов умирают в течение 1 года из-за аспирационной пневмонии в состоянии акинетического мутизма. Используемые в настоящее время диагностические критерии БКЯ включают быстро прогрессирующую деменцию, экстрапирамидно/ пирамидные и зрительные расстройства, миоклонус, мозжечковую атаксию, а также характерные изменения ЭЭГ в виде комплексов высокоамплитудных 2–3-фазных острых волн и положительный маркер белок 14-3-3 в цереброспинальной жидкости (ЦСЖ). Сложности диагностики БКЯ связывают с редкостью заболевания, клиническим полиморфизмом и недостаточной информированностью врачей.
Ряд подтипов спорадического CJD распознается на основе молекулярных маркеров, и они имеют отчетливые клинические и патологические особенности.
- кодон 129 в гене прионного белка (метионин (M) или валин (V))
- размер устойчивого к протеазе ядра аномального прионного белка (PrPSc тип 1 (21 кДа) или тип 2 (19 кДа))
Каждый вариант спорадического CJD является результатом комбинации генотипа кодона 129 (M или V) и типа PrPSc (1, 2 или 1 + 2).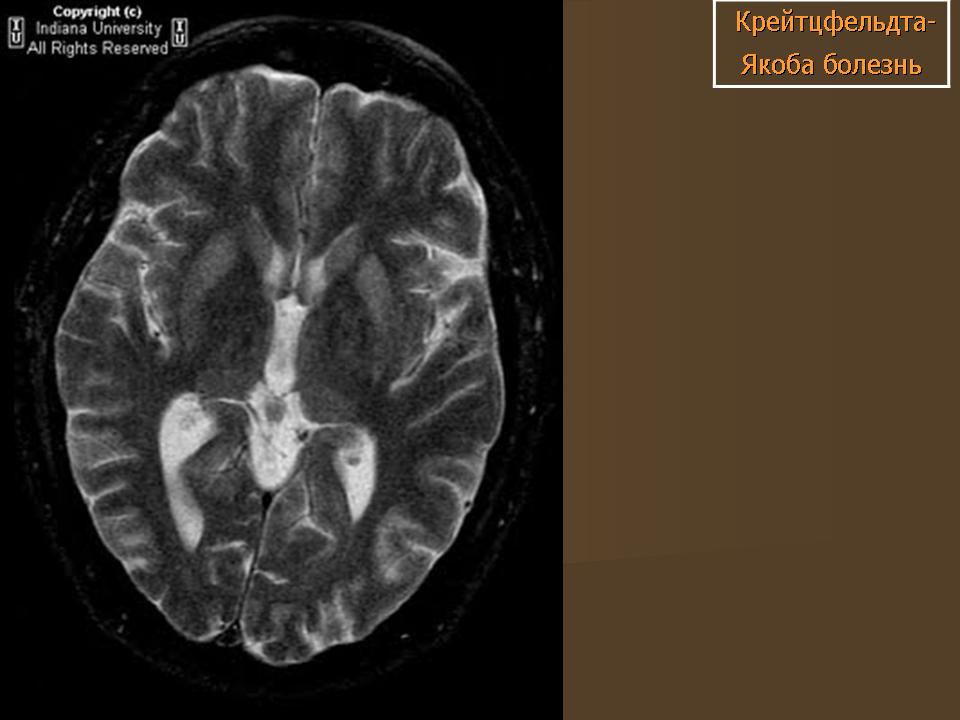 Кроме того, было отмечено, что подтип MM2 имеет отличительные гистопатологические особенности, влияющие на кору головного мозга или таламус, и поэтому был разделен на MM2 кортикальный (MM2C) и MM2 таламический (MM2T). Встречаются также смешанные типы. Результатом является то, что в литературе существуют значительные различия в отношении того, сколько существует подтипов и следует ли и как их группировать вместе.
Кроме того, было отмечено, что подтип MM2 имеет отличительные гистопатологические особенности, влияющие на кору головного мозга или таламус, и поэтому был разделен на MM2 кортикальный (MM2C) и MM2 таламический (MM2T). Встречаются также смешанные типы. Результатом является то, что в литературе существуют значительные различия в отношении того, сколько существует подтипов и следует ли и как их группировать вместе.
Имеется несколько фенотипов заболевания:
- Вариант Хайденхайна спорадической БКЯ является отчетливым клиническим проявлением ряда молекулярных подтипов (MM1 и MM2C), в которых преобладают чисто зрительные нарушения на ранних стадиях заболевания.
- Вариант Браунелла-Оппенгеймера проявляется изолированной мозжечковой атаксией. Результаты ЭЭГ у пациентов с фенотипом Браунелла-Оппенгеймера обычно показывают отсутствие периодических комплексов с резкими волнами (PSWCs). Белок 14-3-3 повышен в анализе ликвора.
- Вариант болезни Крейтцфельдта-Якоба проявляется в основном психиатрическими симптомами (такими как депрессия) и сенсорными симптомами (дизестезии или парестезии).

БКЯ приводит к губкообразной дегенерации головного мозга, которая, как полагают, вызвана превращением нормального прионного белка в белковые инфекционные частицы, которые накапливаются внутри и вокруг нейронов и приводят к гибели клеток. Стоит обратить внимание на следующие особенности:
- типичная картина электроэнцефалографии (ЭЭГ)
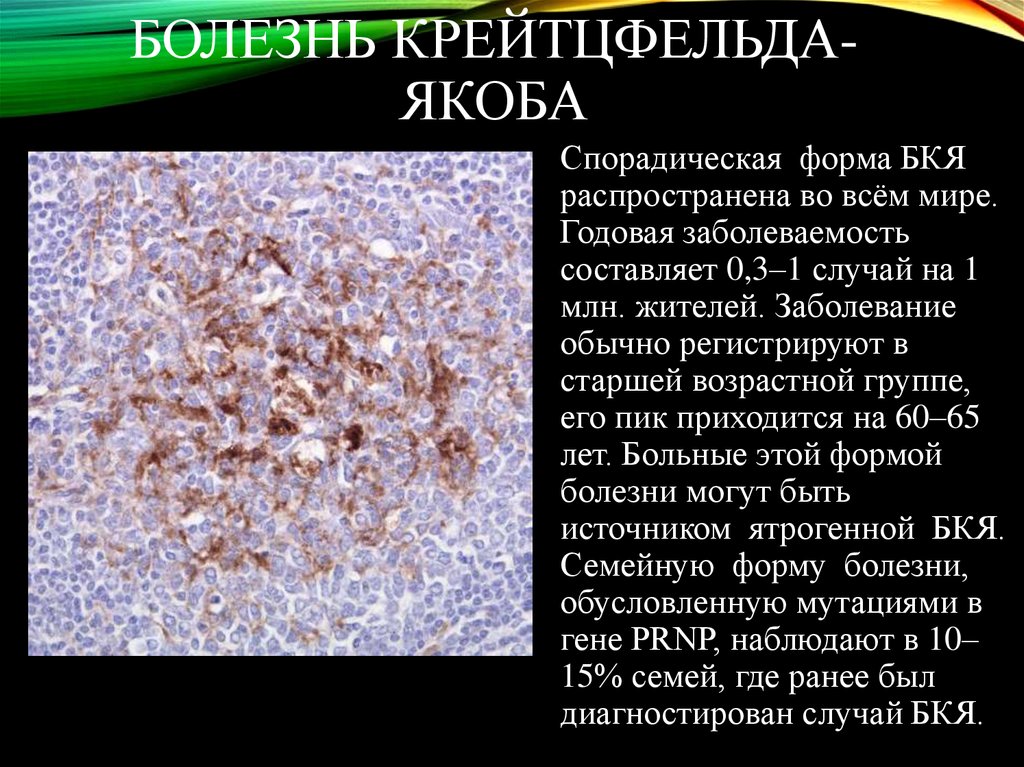
- S100 представляет собой семейство цитоплазматических кальций-связывающих белков, экспрессируемых во многих клеточных линиях, на которые можно нацеливаться с помощью иммуногистохимии. Окрашивание на S100 полезно для характеристики ряда опухолей, включая злокачественную меланому, глиальные опухоли, нейрогенные опухоли (например, шванномы и нейрофибромы), мезенхимальные опухоли (например, хондромы, хондросаркомы, липосаркомы, некоторые остеогенные саркомы) и некоторые гистиоцитарные опухоли
- Белки 14-3-3 выявляются в спинномозговой жидкости, его чувствительность составляет 92%, а его специфичность — 80%.
- Так же в спинномозговой жидкости можно выявить патогмоничного прионного белка, его специфичность выше чем белка 14-3-3.

До недавнего времени «золотым стандартом» верификации диагноза БКЯ являлась биопсия головного мозга, позволяющая выявить характерные изменения в мозговой ткани в виде мелких вакуолей в телах нейронов, из-за чего ткань мозга приобретает губчатый вид, пролиферации клеток глии при отсутствии признаков воспаления. При электронной микроскопии возможно обнаружение прионных палочек, являющихся патогномоничным признаком заболевания. Указанные морфологические изменения отмечаются в коре головного мозга, базальных ганглиях, мозжечке и верхних отделах ствола мозга. Однако в случаях БКЯ биопсия мозга не нашла широкого применения в клинической практике из-за инвазивности метода, сложности санитарной обработки оборудования и утилизации биоматериалов, связанных с высокой устойчивостью прионов, а также вследствие небольшого объема биоптата мозговой ткани, что может быть причиной ложноотрицательных результатов морфологического и иммуногистохимического исследования. Однако в настоящее все больше распространена МРТ диагностика данного заболевания, по причине выявления специфических изменений, наиболее информативными являются последовательность DWI, которая демонстрирует повышенный сигнал более заметный, чем изменения T2 / FLAIR. Сигнальные аномалии при дебюте могут быть незаметными, но становятся более выраженными по мере прогрессирования заболевания.
Однако в настоящее все больше распространена МРТ диагностика данного заболевания, по причине выявления специфических изменений, наиболее информативными являются последовательность DWI, которая демонстрирует повышенный сигнал более заметный, чем изменения T2 / FLAIR. Сигнальные аномалии при дебюте могут быть незаметными, но становятся более выраженными по мере прогрессирования заболевания.
Характеристики МР-сигналов включают в себя следующее:
FLAIR
- Симптом «подушки»: двустороннее симметричное повышение интенсивности сигнала задних отделов таламусов.
- Симптом «хоккейной клюшки»: симметричное повышение интенсивности сигнала от бледных шаров и таламусов.
DWI
- Прогрессирующее повышение интенсивности сигнала от коры больших полушарий гирального характера (симптом «корковой ленты»).
- Изменения сигнала на ДВИ может не выявляться на поздней стадии заболевания.
ADC: вариабельная и зависит от стадии заболевания.
T1ВИ: высокий сигнал от бледного шара.
T1ВИ+C: накопления контраста не отмечается
На ПЭТ с фтором-18-ФДГ выявляют гипометаболизм в пораженных областях.
Возникновение и передача | Классическая болезнь Крейтцфельдта-Якоба (CJD) | Прионная болезнь
Классическая БКЯ известна с начала 1920-х годов. Считается, что большинство случаев CJD (около 85%) возникают спорадически, вызванные спонтанной трансформацией нормальных прионных белков в аномальные прионы. Это спорадическое заболевание встречается во всем мире, включая Соединенные Штаты, со скоростью примерно 1–2 случая на 1 миллион населения в год. Риск CJD увеличивается с возрастом; среднегодовой показатель в 2016–2020 гг. в США составлял около 5 случаев на миллион среди лиц в возрасте 55 лет и старше.
У меньшей части пациентов (5–15%) развивается БКЯ из-за наследственных мутаций гена прионного белка. Эти наследственные формы включают синдром Герстмана-Штраусслера-Шейнкера и фатальную семейную бессонницу. Записи о смерти являются хорошим индикатором заболеваемости CJD, потому что болезнь всегда заканчивается летальным исходом, а средняя продолжительность болезни составляет около 4–5 месяцев.
Записи о смерти являются хорошим индикатором заболеваемости CJD, потому что болезнь всегда заканчивается летальным исходом, а средняя продолжительность болезни составляет около 4–5 месяцев.
*Случаи смерти, полученные на основании данных о множественных причинах смерти, включают все формы прионной болезни человека и основаны на кодах МКБ-9 и МКБ-10 и имеющихся компьютеризированных дословных данных свидетельств о смерти. В эти данные были внесены некоторые изменения на основе соответствующей информации, полученной от других механизмов наблюдения. Ставки приведены к стандартному прогнозируемому населению США на 2000 год.
Подсчет случаев включает лиц с вероятным CJD на основании клинических тестов, таких как положительная конверсия, вызванная землетрясением в реальном времени (RT-QuIC), при условии, что диагноз прионной болезни указан в свидетельстве о смерти. Было показано, что в случае положительного результата тест RT-QuIC убедительно свидетельствует о прионной болезни (Rhoads, et al. ), и, если нет альтернативного диагноза, настоятельно рекомендуется указывать прионную болезнь в свидетельстве о смерти. Ежегодное количество людей с положительными клиническими тестами RT-QuIC, но без анализа ткани головного мозга для подтверждения заболевания, можно найти на веб-сайте Национального центра наблюдения за патологией прионных заболеваний (таблицы NPDPSC).
), и, если нет альтернативного диагноза, настоятельно рекомендуется указывать прионную болезнь в свидетельстве о смерти. Ежегодное количество людей с положительными клиническими тестами RT-QuIC, но без анализа ткани головного мозга для подтверждения заболевания, можно найти на веб-сайте Национального центра наблюдения за патологией прионных заболеваний (таблицы NPDPSC).
| Год | смертей (приблизительно) | Смертность с поправкой на возраст |
|---|---|---|
| 1979 | 179 | 0,850 |
| 1980 | 172 | 0,787 |
| 1981 | 214 | 0,988 |
| 1982 | 201 | 0,901 |
| 1983 | 183 | 0,825 |
| 1984 | 221 | 0,985 |
| 1985 | 235 | 1. 033 033 |
| 1986 | 247 | 1,073 |
| 1987 | 268 | 1,129 |
| 1988 | 243 | 1.020 |
| 1989 | 242 | 1,011 |
| 1990 | 210 | 0,859 |
| 1991 | 238 | 0,982 |
| 1992 | 255 | 1.018 |
| 1993 | 256 | 1.012 |
| 1994 | 278 | 1,079 |
| 1995 | 265 | 1,023 |
| 1996 | 261 | 0,984 |
| 1997 | 305 | 1,152 |
| 1998 | 281 | 1,038 |
| 1999 | 272 | 0,990 |
| 2000 | 238 | 0,853 |
| 2001 | 259 | 0,917 |
| 2002 | 260 | 0,904 |
| 2003 | 284 | 0,967 |
| 2004 | 279 | 0,921 |
| 2005 | 296 | 0,972 |
| 2006 | 290 | 0,925 |
| 2007 | 330 | 1. 019 019 |
| 2008 | 352 | 1.086 |
| 2009 | 353 | 1,067 |
| 2010 | 396 | 1,149 |
| 2011 | 409 | 1.161 |
| 2012 | 380 | 1.054 |
| 2013 | 478 | 1.300 |
| 2014 | 441 | 1,168 |
| 2015 | 481 | 1,227 |
| 2016 | 492 | 1,225 |
| 2017 | 510 | 1.241 |
| 2018 | 479 | 1.141 |
| 2019 | 561 | 1,319 |
| 2020 | 538 | 1,235 |
Инфекционный контроль | Классическая болезнь Крейтцфельдта-Якоба (CJD) | Прионная болезнь
- Ятрогенная передача БКЯ
- Обработка хирургических инструментов, используемых у пациентов с подозрением или подтвержденным диагнозом БКЯ
- Методы автоклавной стерилизации, описанные в Руководстве ВОЗ
- Обработка инструментов, используемых у пациентов с неясным диагнозом БКЯ
- Деконтаминация термочувствительных инструментов или материалов
- Меры предосторожности при бальзамировании тел пациентов с подозрением или подтвержденным диагнозом БКЯ
- Ресурс
 Эти случаи были связаны с использованием зараженного гормона роста человека, трансплантатов твердой мозговой оболочки и роговицы или нейрохирургического оборудования. Из шести случаев, связанных с использованием зараженного оборудования, четыре были связаны с нейрохирургическими инструментами и два со стереотаксическими глубинными электродами ЭЭГ.
Эти случаи были связаны с использованием зараженного гормона роста человека, трансплантатов твердой мозговой оболочки и роговицы или нейрохирургического оборудования. Из шести случаев, связанных с использованием зараженного оборудования, четыре были связаны с нейрохирургическими инструментами и два со стереотаксическими глубинными электродами ЭЭГ.Все эти случаи, связанные с оборудованием, произошли до рутинного внедрения процедур стерилизации, используемых в настоящее время в медицинских учреждениях. О таких случаях не сообщалось с 1976 г., и не было выявлено ни одного ятрогенного случая БКЯ, связанного с воздействием агента БКЯ с таких поверхностей, как полы, стены или столешницы.
Обработка хирургических инструментов, используемых у пациентов с подозрением или подтвержденным диагнозом CJD
В исследованиях инактивации не проводилась строгая оценка эффективности фактических методов очистки и обработки, используемых в медицинских учреждениях. Рекомендации по обработке инструментов, потенциально зараженных агентом CJD, в первую очередь основаны на исследованиях инактивации in vitro, в которых использовались либо ткани головного мозга, либо гомогенаты тканей, оба из которых создают огромные проблемы для любого процесса стерилизации.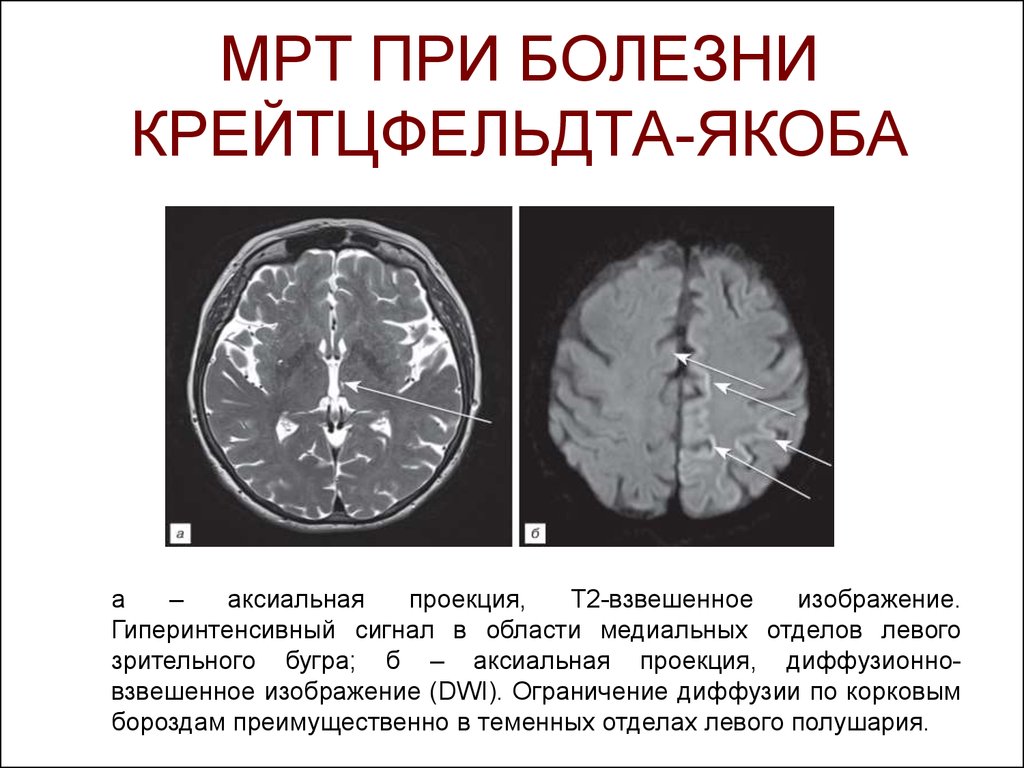
Всемирная организация здравоохранения (ВОЗ) разработала руководство по инфекционному контролю БКЯ, которое может быть ценным руководством для сотрудников инфекционного контроля и других медицинских работников, занимающихся лечением пациентов с БКЯ. Уничтожение термостойких хирургических инструментов, соприкасающихся с тканями с высокой инфекционностью, хотя и является самым безопасным и однозначным методом, описанным в рекомендациях ВОЗ, может оказаться непрактичным или экономически неэффективным.
Один из трех наиболее строгих методов химической и автоклавной стерилизации, изложенных в Приложении III руководства ВОЗ (см. ниже), следует использовать для обработки термостойких инструментов, контактирующих с тканями пациентов с подозрением или подтвержденным заболеванием CJD. Инструменты следует поддерживать во влажном состоянии и не допускать их высыхания на воздухе на протяжении всей хирургической процедуры путем погружения их в воду или дезинфицирующий раствор.
Химические и автоклавные методы стерилизации, описанные в Приложении III к Руководству ВОЗ по инфекционному контролю при трансмиссивных губчатых энцефалопатиях
Ниже перечислены три наиболее строгих метода стерилизации термостойких инструментов, описанных в Приложении III к Руководству ВОЗ; методы перечислены в порядке от более к менее серьезному лечению. Гипохлорит натрия может вызвать коррозию некоторых инструментов, например инструментов с золотым покрытием. Перед погружением инструментов в гипохлорит натрия следует проконсультироваться с изготовителем инструментов о переносимости инструмента при воздействии гипохлорита натрия. Инструменты должны быть обеззаражены с помощью комбинации химических и рекомендуемых методов автоклавирования, прежде чем подвергать их очистке в цикле мойки и обычной стерилизации.
Гипохлорит натрия может вызвать коррозию некоторых инструментов, например инструментов с золотым покрытием. Перед погружением инструментов в гипохлорит натрия следует проконсультироваться с изготовителем инструментов о переносимости инструмента при воздействии гипохлорита натрия. Инструменты должны быть обеззаражены с помощью комбинации химических и рекомендуемых методов автоклавирования, прежде чем подвергать их очистке в цикле мойки и обычной стерилизации.
- Погрузить в кастрюлю, содержащую 1 н. раствор гидроксида натрия (NaOH), и нагревать в автоклаве с гравитационным вытеснением при 121°C в течение 30 мин; чистый; промыть в воде; и подлежат плановой стерилизации.
[ПРИМЕЧАНИЕ CDC: поддон с гидроксидом натрия должен быть накрыт, и следует соблюдать осторожность, чтобы не допустить разлива гидроксида натрия в автоклаве. Во избежание воздействия в автоклаве газообразного гидроксида натрия, конденсирующегося на крышке контейнера, рекомендуется использовать контейнеры с ободком и крышкой, предназначенными для сбора конденсата и его стекания обратно в поддон. Лица, использующие эту процедуру, должны соблюдать осторожность при работе с горячим раствором гидроксида натрия (после автоклавирования) и избегать потенциального воздействия газообразного гидроксида натрия, проявлять осторожность на всех этапах стерилизации и давать автоклаву, инструментам и растворам остыть перед извлечением. . Значок эксперимента в формате pdf [PDF – 88 КБ], проведенный исследователями Управления по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA), показал, что использование соответствующих поддонов и крышек предотвращает утечку паров гидроксида натрия, которые могут повредить автоклав (Brown and Merritt. Am J Infect Контроль 2003;31:257-260).] - Погрузить в 1 н. раствор NaOH или гипохлорит натрия (20 000 частей на миллион доступного хлора) на 1 час; переносить инструменты в воду; нагревание в автоклаве с гравитационным вытеснением при 121°C в течение 1 часа; чистый; и подлежат плановой стерилизации.
- Погрузить в 1 н. раствор NaOH или гипохлорит натрия (20 000 частей на миллион доступного хлора) на 1 час; выньте и промойте в воде, а затем переложите в открытый лоток и нагревайте в автоклаве с гравитационным вытеснением (121°С) или с пористой загрузкой (134°С) в течение 1 часа; чистый; и подлежат плановой стерилизации.
[ПРИМЕЧАНИЕ CDC: гипохлорит натрия может вызывать коррозию некоторых инструментов.]
 Лица, использующие эту процедуру, должны соблюдать осторожность при работе с горячим раствором гидроксида натрия (после автоклавирования) и избегать потенциального воздействия газообразного гидроксида натрия, проявлять осторожность на всех этапах стерилизации и давать автоклаву, инструментам и растворам остыть перед извлечением. . Значок эксперимента в формате pdf [PDF – 88 КБ], проведенный исследователями Управления по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA), показал, что использование соответствующих поддонов и крышек предотвращает утечку паров гидроксида натрия, которые могут повредить автоклав (Brown and Merritt. Am J Infect Контроль 2003;31:257-260).]
Лица, использующие эту процедуру, должны соблюдать осторожность при работе с горячим раствором гидроксида натрия (после автоклавирования) и избегать потенциального воздействия газообразного гидроксида натрия, проявлять осторожность на всех этапах стерилизации и давать автоклаву, инструментам и растворам остыть перед извлечением. . Значок эксперимента в формате pdf [PDF – 88 КБ], проведенный исследователями Управления по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA), показал, что использование соответствующих поддонов и крышек предотвращает утечку паров гидроксида натрия, которые могут повредить автоклав (Brown and Merritt. Am J Infect Контроль 2003;31:257-260).] ]
] Исследователи Управления по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) оценили влияние на хирургические инструменты шагов, включенных в протоколы стерилизации, перечисленные выше; некоторые из протоколов, которые они оценили, подвергали инструменты более жестким условиям, чем предписанные выше. Их результаты показывают, что большая часть повреждений от автоклавирования в гидроксиде натрия была косметической и не повлияла на работу или очистку инструментов. Замачивание в гидроксиде натрия оказало наименьшее повреждающее воздействие на инструменты, но погружение в отбеливатель гипохлорита натрия вызвало серьезные повреждения некоторых инструментов.
Обработка инструментов, используемых у пациентов с неясным диагнозом БКЯ во время нейрохирургической процедуры
У некоторых пациентов, подвергающихся нейрохирургическим вмешательствам, диагноз БКЯ, не подозревавшийся до процедуры, может быть подтвержден после нейрохирургической операции. Для этой группы пациентов, у которых клинический диагноз, ведущий к нейрохирургическому вмешательству, остается неясным, инструменты следует обрабатывать таким же образом, как инструменты, используемые в процедурах у пациентов с подозрением или подтвержденным диагнозом CJD. Если не установлен четкий диагноз, не связанный с CJD, этих пациентов следует рассматривать как потенциально подозреваемых пациентов с CJD для всех других требований инфекционного контроля.
Обеззараживание термочувствительных инструментов или материалов, контактировавших с пациентами с подозрением или подтвержденным диагнозом CJD
Все одноразовые инструменты, материалы и отходы, контактирующие с тканями с высокой инфекционностью (головной, спинной мозг и глаза) и тканями с низкой инфекционностью (спинномозговая жидкость, почки, печень, легкие, лимфатические узлы, селезенка и плацента) пациентов с подозрением или подтвержденным ТГЭ следует утилизировать путем сжигания. Поверхности и термочувствительные инструменты многоразового использования, которые соприкасаются с тканями с высокой и низкой инфекционностью, следует дезинфицировать путем замачивания или замачивания в 2 н. растворе NaOH или неразбавленного гипохлорита натрия в течение 1 часа и промывания водой.
Поверхности и термочувствительные инструменты многоразового использования, которые соприкасаются с тканями с высокой и низкой инфекционностью, следует дезинфицировать путем замачивания или замачивания в 2 н. растворе NaOH или неразбавленного гипохлорита натрия в течение 1 часа и промывания водой.
[ПРИМЕЧАНИЕ CDC: гипохлорит натрия может вызывать коррозию некоторых инструментов.]
Меры предосторожности при бальзамировании тел пациентов с подозрением или подтвержденным диагнозом CJD
Вскрытое или травмированное тело пациента с подозрением или подтвержденным диагнозом CJD может быть забальзамировано с соблюдением мер предосторожности изложено в Руководстве ВОЗ по инфекционному контролю CJDвнешний значок. Пациентов с CJD, которые не подвергались вскрытию или тела которых не были травмированы, можно бальзамировать с соблюдением стандартных мер предосторожности. Родственникам пациентов с БКЯ следует рекомендовать избегать поверхностных контактов (таких как прикосновения или поцелуи лица пациента) с телом пациента с БКЯ, прошедшего аутопсию.