
Миотония томпсона: Миотония Томсена. Что такое Миотония Томсена?
Миотония Томсена. Что такое Миотония Томсена?
ВАЖНО
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Миотония Томсена — наследственно-семейное поражение поперечно-полосатой мускулатуры, проявляющееся пролонгированным расслаблением мышц после их сокращения. Кроме типичного миотонического феномена, заболевание характеризуется гипертрофическими изменениями пораженных мышц, манифестацией с поражения кистей, частой вовлеченностью лицевой мускулатуры. Миотония Томсена диагностируется на основании анамнестических данных, результатов неврологического осмотра, ЭМГ и ЭНГ, молекулярно-генетического исследования. Лечение сводится к назначению препаратов, уменьшающих тонические реакции мышц и нормализующих ионное состояние миофибрилл.
- Этиология и патогенез миотонии Томсена
- Симптомы миотонии Томсена
- Диагностика миотонии Томсена
- Лечение миотонии Томсена
- Прогноз и профилактика
- Цены на лечение
Общие сведения
Миотония Томсена входит в группу наследственных миотоний, включающую также миотонию Россолимо-Штейнерта-Куршмана, врожденную парамиотонию Эйленбурга, миотонию Беккера и еще ряд заболеваний. Болезнь Томсена приобрела свое название в соответствии с фамилией ученого, подробно описавшего ее симптомы и порядок наследования на примере 4-х поколений своей семьи. За 2 года до Томсена (в 1874 г.) первое описание заболевания дал Лейден. Поэтому в современной неврологии употребляется также название болезнь Лейдена-Томсена.
У больных с данным видом миотонии прослеживается доминантный тип наследования патологического аутосомного гена. Частота встречаемости заболевания составляет по различным оценкам от 3 до 7 человек на 1 млн.
Миотония Томсена
Этиология и патогенез миотонии Томсена
Миотония Томсена относится к наследственным каналопатиям. Как и миотония Беккера, заболевание связано с дефектом 7-й хромосомы, а именно гена CLCN1, детерминирующего синтез белка хлорных ионных каналов миофибрилл скелетной мускулатуры. Следствием нарушения синтеза этого специфического белка является уменьшенное прохождение ионов хлора внутрь миофибриллы и их скопление на поверхности мембраны мышечного волокна (сарколеммы). Возникающая в результате биоэлектрическая нестабильность сарколеммы обуславливает ее чрезмерную возбудимость. При этом периферический неврон функционирует без отклонений, но на обычный нервный импульс мембрана миофибриллы реагирует повышенным возбуждением, которое препятствует нормальному расслаблению мышечного волокна после его сокращения. Причем, чем быстрее происходит сокращение миофибриллы, тем более затрудненно ее расслабление.
Морфологические изменения мышечной ткани неспецифичны для миотонии Томсена и являются типичными для большинства миотоний. Отмечается централизация ядер сарколеммы, увеличение площади сечения миофибрилл, свидетельствующее о их гипертрофии. Электронная микроскопия определяет гипертрофию саркоплазматического ретикулума, увеличение размеров митохондрий и изменение их формы, утолщение телофрагмы.
Симптомы миотонии Томсена
В большинстве случаев манифестация болезни приходится на ранний детский возраст. Ведущим в клинике заболевания является миотонический симптомокомплекс, так называемый миотонический феномен. Он характеризуется пролонгированной мышечной релаксацией после совершения быстрого движения. Клинически это проявляется невозможностью быстро произвести следующее движение из-за тонического спазма мышц, участвовавших в предыдущем двигательном акте.
Патогномоничным для болезни Томсена является ее манифестация с патологических изменений в кистях. Затем миотонические проявления распространяются на мускулатуру ног, мимическую и жевательную группы мышц. В результате тонические спазмы наблюдаются при резком начале ходьбы, зажмуривании глаз, смыкании зубов и т. п. движениях. При попытке быстрой ходьбы пациенты утрачивают равновесие и могут упасть из-за развития общей скованности. Эмоциональные переживания и пребывание в холоде усиливают миотонические реакции.
Своеобразен внешний вид пациентов с миотонией Томсена. Вследствие диффузной мышечной гипертрофии они зачастую имеют телосложение атлетов. Их мышцы отличаются чрезмерно плотной консистенцией, но имеют сниженную силу. Мышечная гипертрофия является отличительным симптомом заболевания, позволяющим дифференцировать его от миотонической дистрофии Россолимо-Штейнерта-Куршмана, сопровождающейся атрофиями мышц.
Мышечная гипертрофия является отличительным симптомом заболевания, позволяющим дифференцировать его от миотонической дистрофии Россолимо-Штейнерта-Куршмана, сопровождающейся атрофиями мышц.
Диагностика миотонии Томсена
Поскольку миотония Томсена обычно манифестирует в раннем возрасте, родители заболевшего ребенка обращаются прежде всего к педиатру, который и направляет их на консультацию детского невролога. Для выявления миотонического симптомокомплекса невролог просит обследуемого несколько раз быстро сжать и разжать кулак. Замедленное разжимание пальцев в начале тестирования и постепенная нормализация движений при их повторении свидетельствуют в пользу миотонии. О наличие миотонического феномена говорит образование «валика» локального мышечного сокращения в ответ на постукивание неврологическим молоточком по мышце. В неврологическом статусе выявляется снижение мышечной силы и миотонический характер сухожильных рефлексов. Возможна миотоническая реакция зрачков на световое раздражение.
Основополагающими в диагностике миотонии Томсена являются электрофизиологические исследования: электромиография и электронейрография. Первая выявляет специфичные для миотонии повторяющиеся высокочастотные разряды, а вторая дает возможность полностью исключить поражение анимальной нервной системы.
Биопсия мышц позволяет изучить морфологические изменения мышечной ткани. Тем не менее, выявляемая при болезни Томсена гипертрофия миофибрилл и централизация ядер их сарколеммы наблюдается и при других типах миотоний. Для постановки точного диагноза с верификацией вида миотонии необходимо проведение молекулярно-генетического анализа, позволяющего определить дефект гена CLCN1.
Лечение миотонии Томсена
Современной медициной еще не разработана радикальная терапия генных заболеваний. Поэтому основная задача лечения миотонии Томсена сводится к уменьшению ее проявлений и замедлению прогрессирования симптомов. Для снижения тонической спастичности используются такие препараты, как дифенин, фенитоин, карбамазепин.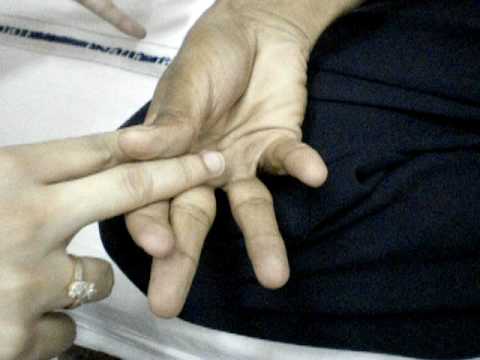 Для поддержания ионного равновесия мембран миофибрилл применяют препараты кальция в виде медикаментозной терапии, электрофореза, гальванизации воротниковой зоны. С этой же целью рекомендованы мероприятия по уменьшению содержания калия в организме: диета с малым содержанием калия, мочегонная терапия. Из мочегонных средств предпочтительно назначение ацетазоламида, поскольку, кроме усиленного выведения калия с мочой, он также увеличивает проницаемость сарколеммы.
Для поддержания ионного равновесия мембран миофибрилл применяют препараты кальция в виде медикаментозной терапии, электрофореза, гальванизации воротниковой зоны. С этой же целью рекомендованы мероприятия по уменьшению содержания калия в организме: диета с малым содержанием калия, мочегонная терапия. Из мочегонных средств предпочтительно назначение ацетазоламида, поскольку, кроме усиленного выведения калия с мочой, он также увеличивает проницаемость сарколеммы.
Особое значение в комплексном лечении миотонии Томсена имеют массаж и ЛФК. Они должны проводиться с особой осторожностью, чтобы улучшить обменные процессы мышечной ткани и одновременно уменьшить ее наклонность к тоническому спазмированию.
Прогноз и профилактика
Миотония Томсена сохраняется в течение всей жизни. Доброкачественное течение и медленное прогрессирование болезни делает ее прогноз относительно благоприятным. Пациенты, как правило, сохраняют трудоспособность. Однако, их профессия не должна быть связана с необходимостью совершать быстрые движения.
К мерам первичной профилактики болезни Томсена можно отнести консультирование генетиком семейной пары, планирующей беременность и имеющей случаи заболевания среди родственников. Профилактикой миотонических приступов служит исключение резкой двигательной активности, переохлаждения, физических перегрузок и эмоционального волнения.
Источники
- Настоящая статья подготовлена по материалам сайта: https://www.krasotaimedicina.ru/
ВАЖНО
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Миотония Томсена/Беккера, CLCN1 ч.м. — узнать цены на анализ и сдать в Москве
Метод определения ПЦР, секвенированиеИсследуемый материал Цельная кровь (с ЭДТА)
Доступен выезд на дом
Исследование частых мутаций в гене CLCN1.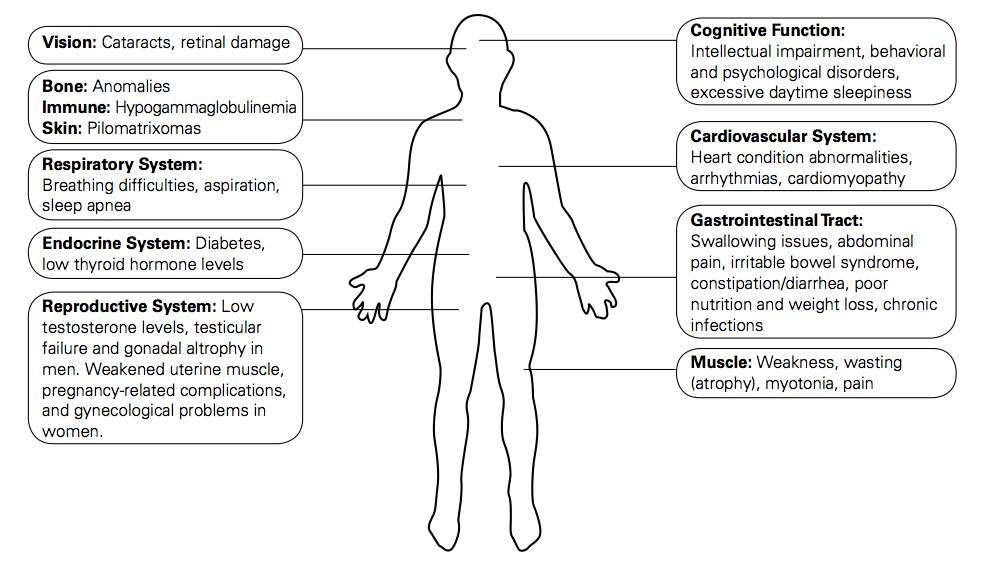
Тип наследования.
Аутосомно-доминантный, аутосомно-рецессивный.
Гены, ответственные за развитие заболевания.
CLСN1 (CHLORIDE CHANNEL 1, SKELETAL MUSCLE) – ген белка мышечных хлорных каналов, расположен на хромосоме 7 в регионе 7q35, содержит 23 экзона.
Определение заболевания.
Миотония — нервно-мышечное заболевание, характеризующееся наличием мышечной гипертрофии и миотонического феномена — замедленной релаксацией мышцы после ее сокращения (сократившаяся мышца долгое время не расслабляется и затем расслабление происходит крайне медленно).
Патогенез и клиническая картина.
Заболевание встречается в двух вариантах — миотонии Томсена (OMIM: 160800) с аутосомно-доминантным типом наследования и миотонии Беккера (OMIM: 255700) с аутосомно-рецессивным типом наследования. Показано, что заболевания являются аллельными вариантами и эффект доминантности и рецессивности обусловлен различными мутациями в одном и том же гене. Миотония Беккера характеризуются ранней манифестацией и более тяжёлыми клиническими проявлениями Белок мышечных хлорных каналов регулирует электрическую возбудимость мембраны скелетных мышц.
1 вариант — миотония Томсена
Первые симптомы заболевания можно отметить с рождения или в периоде новорожденности. Основным проявлением заболевания является миотонический спазм в различных группах мышц, возникающий после их интенсивного произвольного сокращения. Выраженность спазма наибольшая в начале движения и уменьшается во время повторных мышечных сокращений. Усиление миотонического феномена возникает на холоде, уменьшение — в тепле, во время отдыха и при приеме небольших доз алкоголя. Наиболее часто первые признаки заболевания возникают в дистальных отделах рук. По мере прогрессирования заболевания в патологический процесс вовлекаются мышцы ног, а также жевательная и мимическая мускулатура. Характерно возникновение перкуссионных миотонических феноменов (миотонического валика и ямки). В ряде случаев, особенно при попытке произвести быстрое движение у больных возникает генерализованный миотонический спазм, во время которого может произойти резкое падение больного, сопровождающееся общей скованностью. Мышечная система больных обычно гипертрофирована, и больные имеют вид атлетически сложенных людей. При этом мышечная сила может быть снижена. Течение заболевания обычно доброкачественное. В ряде случаев отмечается слабое прогрессирование заболевания.
Мышечная система больных обычно гипертрофирована, и больные имеют вид атлетически сложенных людей. При этом мышечная сила может быть снижена. Течение заболевания обычно доброкачественное. В ряде случаев отмечается слабое прогрессирование заболевания.
2 вариант — миотония Беккера
В большинстве случаев заболевание возникает в возрасте от 4 до 12 лет у девочек и около 18 лет у мальчиков. Первые признаки миотонии возникают в мышцах ног, через несколько лет — в мышцах рук. У некоторых больных в поздних стадиях заболевания отмечено вовлечение в процесс лицевой мускулатуры. Клинические проявления заболевания сходны с таковыми при миотонии Томсена, однако, более выражены и у части больных сопровождаются миалгией и слабостью проксимальных групп мышц. Мышечные гипертрофии встречаются редко.
На электромиограмме выявляется специфический миотонический феномен.
У части больных может быть нерезко выраженное увеличение уровня активности креатинфосфокиназы в плазме крови.
Специфические морфологические признаки отсутствуют.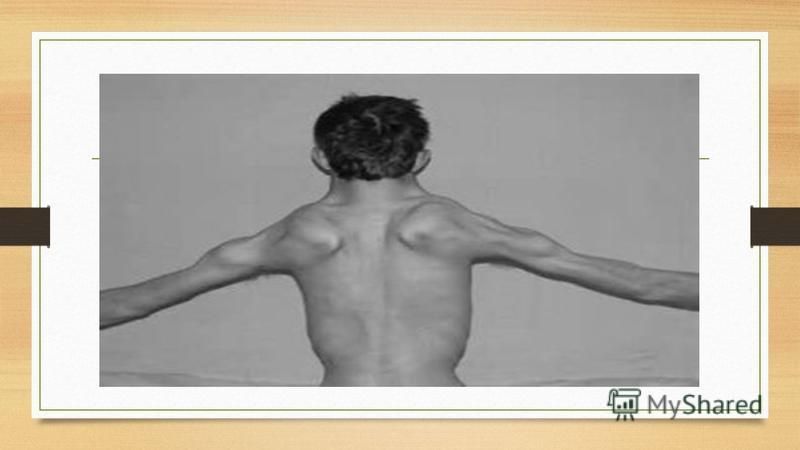 В большинстве случаев выявляется вариабельный диаметр мышечных волокон, их гипертрофия и централизация ядер.
В большинстве случаев выявляется вариабельный диаметр мышечных волокон, их гипертрофия и централизация ядер.
Частота встречаемости: в Европе оценивается как 1:100 000 семей, в странах Скандинавии – 1:10 000.
Перечень исследуемых мутаций может быть предоставлен по запросу.
Врожденная миотония (болезнь Томсена и тип Беккера) — Болезни
Перейти к основному содержанию
- О нас
Присоединиться к MDA
Место, где люди и семьи находятся в центре всего, что мы делаем.
- О МДА
- Наше влияние
- История MDA
- Часто задаваемые вопросы
- МДА Лидерство
- Национальные послы
- Карьера
- Пресс-центр
- Финансы
- Журнал квестов
- Свяжитесь с нами
- нервно-мышечные заболевания
Присоединиться к MDA
Место, где люди и семьи находятся в центре всего, что мы делаем.

- Нервно-мышечные заболевания
- Полный список болезней
- Боковой амиотрофический склероз (БАС)
- Болезнь Шарко-Мари-Тута (ШМТ)
- Врожденная мышечная дистрофия (ВМД)
- Мышечная дистрофия Дюшенна (МДД)
- Мышечная дистрофия Эмери-Дрейфуса
- Эндокринные миопатии
- Метаболические заболевания мышц
- Митохондриальные миопатии (ММ)
- Миотоническая дистрофия (DM)
- Спинально-бульбарная мышечная атрофия (СБМА)
- Спинальная мышечная атрофия (СМА)
- Уход и услуги
Присоединиться к MDA
Место, где люди и семьи находятся в центре всего, что мы делаем.
- Об услугах MDA
- Медицинские центры MDA
- Познакомьтесь со своей командой
- Ваш визит
- Общественное образование MDA
- Учебные материалы
- Доступ к мастерским
- Вебинары по запросу
- Национальный ресурсный центр
- MDA Connect Назначения
- Запрос информации
- Ресурсы сообщества
- Программы и информация сторонних организаций
- Летний лагерь MDA
- Стать волонтером лагеря
- Подать заявку в лагерь
- Виртуальный лагерь
- Наука и исследования
Присоединиться к MDA
Место, где люди и семьи находятся в центре всего, что мы делаем.
- Об исследованиях MDA
- Краткий обзор грантов
- Создание новой терапии
- Чего мы достигли
- Концентратор данных MOVR
- Средство поиска клинических испытаний
- Наша исследовательская программа
- Возможности финансирования
- Свяжитесь с нашей исследовательской группой
- Ежегодная конференция MDA
- Венчурная благотворительность MDA
- Программа быстрого запуска MDA
- Средство поиска клинических испытаний
- Ежегодная конференция MDA
- Медицинское образование
- Медицинские работники и исследователи Подпишитесь на рассылку новостей
- Способы дать
Апрель — месяц добровольцев
Отметьте вместе с нами наших щедрых добровольцев во время Национального месяца добровольцев.
- Пожертвовать сейчас
- Сделать ежемесячный подарок
- Пожертвовать в честь или память
- Подходящие подарки
- Пожертвовать акции
- Фонды, рекомендованные донорами
- Пожертвовать криптовалюту
- Оставить наследие
- Подари, пока покупаешь
- Втягиваться
Апрель — 9-й месяц волонтерства0003
Отметьте вместе с нами наших щедрых добровольцев во время Национального месяца добровольцев.
- Стать волонтером
- Волонтерские ресурсы
- Стать адвокатом
- Принять участие в событии
- Виртуальные события
- Начать сбор средств
- Стать партнером MDA
- Познакомьтесь с нашими партнерами
- Пожарные для MDA
- Присоединяйтесь к МДА
- Квест Медиа
Присоединиться к MDA
Место, где люди и семьи находятся в центре всего, что мы делаем.
- Журнал
- Информационный бюллетень
- Подкаст
- Блог




карта Найти заботу Поиск Поиск Пожертвовать
Врожденная миотония (болезнь Томсена и тип Беккера)
Что такое врожденная миотония?
Врожденная миотония присутствует с раннего детства, но симптомы могут быть легкими. В зависимости от формы расстройства симптомы и признаки могут проявляться в период от младенчества до 2–3 лет при синдроме Томсена и в возрасте от 4 до 12 лет при синдроме Беккера. Большинство людей с врожденной миотонией ведут долгую и продуктивную жизнь. Хотя ригидность мышц может мешать повседневной деятельности (т. е. ходьбе, хватанию, жеванию и глотанию), она обычно уменьшается при физических нагрузках. Статью о профессиональном боксере с врожденной миотонией см. Несмотря на болезнь мышц, «Игрушечный тигр» был потрясающей боевой машиной.
Статью о профессиональном боксере с врожденной миотонией см. Несмотря на болезнь мышц, «Игрушечный тигр» был потрясающей боевой машиной.
Существует два типа врожденной миотонии: миотония типа Беккера — наиболее распространенная форма, а болезнь Томсена — очень редкая, относительно легкая форма.
Каковы симптомы врожденной миотонии?
Основными проблемами, с которыми сталкиваются люди с этим заболеванием, являются замедленное расслабление мышц и ригидность мышц, обычно провоцируемые резкими движениями после отдыха. См. Признаки и симптомы.
Что вызывает врожденную миотонию?
Это заболевание вызывается мутациями в гене хлоридного канала, необходимого для отключения электрического возбуждения, вызывающего сокращение мышц.
Тип Беккера наследуется по аутосомно-рецессивному типу , что означает, что он продуцируется дефектными генами, переданными обоими родителями.
Тип Томсена является аутосомно-доминантным , что означает, что он производится дефектным геном, внесенным одним из родителей. Дополнительную информацию см. в разделе «Причины/наследование».
Дополнительную информацию см. в разделе «Причины/наследование».
Как проявляется врожденная миотония?
Врожденная миотония начинается в раннем или позднем детстве и не прогрессирует. Человек с врожденной миотонией может прожить долгую и продуктивную жизнь и даже преуспеть в спорте, где сила важнее ловкости.
Статью о профессиональном боксере с врожденной миотонией см. в разделе «Несмотря на болезнь мышц, игрушечный тигр был потрясающей боевой машиной».
Каков статус исследований врожденной миотонии?
MDA поддерживает текущие исследования молекулярных основ наследственных миопатий и поиск эффективных методов лечения. Дополнительную информацию см. в разделе Исследования.
Найдите MDA
в своем сообществе
Штат или почтовый индекс
Врожденная миотония — StatPearls — NCBI Bookshelf
Непрерывное обучение
Врожденная миотония (ВК) — это генетическая нервно-мышечная каналопатия, которая обычно проявляется в раннем детстве и приводит к задержке расслабления скелетных мышц после сокращения.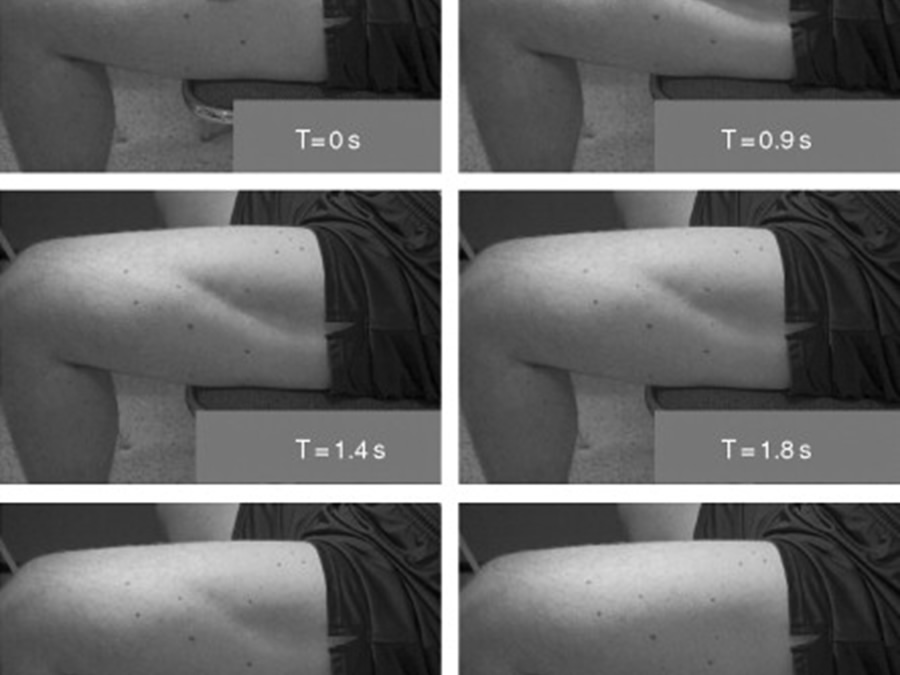 СК имеет множество последствий для пациентов и их семей, включая нарушение движения, глотания, желудочно-кишечные расстройства и респираторные осложнения. В этом упражнении описывается представление, оценка и диагностика СК, а также подчеркивается роль межпрофессиональной команды в уходе за пациентами с этим заболеванием.
СК имеет множество последствий для пациентов и их семей, включая нарушение движения, глотания, желудочно-кишечные расстройства и респираторные осложнения. В этом упражнении описывается представление, оценка и диагностика СК, а также подчеркивается роль межпрофессиональной команды в уходе за пациентами с этим заболеванием.
Цели:
Изучите этиологию врожденной миотонии.
Опишите соответствующий процесс оценки врожденной миотонии.
Опишите текущие варианты лечения врожденной миотонии.
Кратко опишите важность сотрудничества и коммуникации между межпрофессиональной командой для улучшения координации помощи пациентам с врожденной миотонией.
Получите доступ к бесплатным вопросам с несколькими вариантами ответов по этой теме.
Введение
Врожденная миотония (ВК) представляет собой генетическую нервно-мышечную каналопатию, поражающую волокна скелетных мышц (полосатые мышцы, контролируемые соматической нервной системой).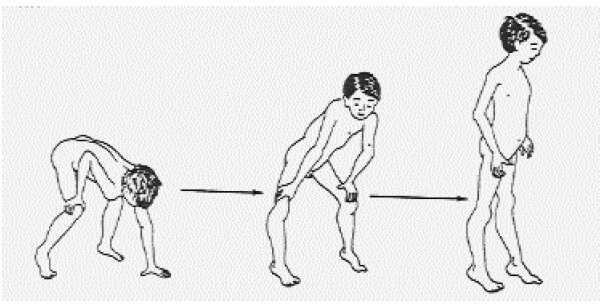 [1] Миотония, определяемая как задержка или нарушение расслабления сокращенных скелетных мышц, считается отличительной чертой заболевания и приводит к длительной ригидности, приводящей к скованности, судорогам и мышечной гипертрофии. СК вызывается мутациями в гене CLCN1, кодирующем потенциалзависимые хлоридные каналы (CIC-1) в мембране сарколеммы.[2] Дефектные каналы CIC-1 вызывают неадекватную повышенную возбудимость клеток скелетных мышц, что приводит к повторяющейся деполяризации и миотонии.[3]
[1] Миотония, определяемая как задержка или нарушение расслабления сокращенных скелетных мышц, считается отличительной чертой заболевания и приводит к длительной ригидности, приводящей к скованности, судорогам и мышечной гипертрофии. СК вызывается мутациями в гене CLCN1, кодирующем потенциалзависимые хлоридные каналы (CIC-1) в мембране сарколеммы.[2] Дефектные каналы CIC-1 вызывают неадекватную повышенную возбудимость клеток скелетных мышц, что приводит к повторяющейся деполяризации и миотонии.[3]
Исторически СК делили на два отдельных состояния: болезнь Беккера и болезнь Томсена. Болезнь Беккера (ББ) наследуется по аутосомно-рецессивному типу и обычно приводит к более тяжелой миотонической картине, которая может прогрессировать до постоянной слабости. Болезнь Томсена (ТБ) является аутосомно-доминантным заболеванием, проявляющимся в детстве раньше, чем ББ, и может быть связана с более легкими проявлениями.[4] Усовершенствованные методы секвенирования позволили идентифицировать более 200 патогенных мутаций в гене CLCN1. [5] По мере увеличения знаний о разнообразии этих мутаций и их фенотипов классификация СК стала менее отчетливой и более разнообразной.
[5] По мере увеличения знаний о разнообразии этих мутаций и их фенотипов классификация СК стала менее отчетливой и более разнообразной.
Этиология
Миотония при СК является результатом дефекта потенциалзависимого хлоридного канала скелетных мышц (CIC-1), вторичного по отношению к мутациям в гене CLCN1.[6] Эти анионные хлоридные каналы многочисленны по всей клеточной мембране и необходимы для поддержания нормальной клеточной возбудимости, а также для высвобождения нейротрансмиттеров и транспорта ионов.[7] Образующийся в результате дефект вызывает повышенную возбудимость мышечной оболочки.[1] Неисправные каналы CIC-1 подавляют проводимость ионов хлорида к сарколеммальной мембране. Это приводит к нарушению способности скелетных мышц поддерживать физиологическую возбудимость. Каналы CIC-1 обеспечивают 80% проводимости мышечной мембраны в состоянии покоя. Поэтому, когда это теряется из-за мутаций MC, входное сопротивление скелетных мышц значительно увеличивается.
Каналы CIC-1 также противодействуют деполяризующему эффекту чрезмерного накопления ионов калия во время возбуждения множественных потенциалов действия (ПД) во внеклеточном пространстве скелетных мышц. У здоровых людей высокая концентрация ионов калия нивелируется конкордантностью каналов CIC-1. Когда этот эффект утрачивается, как при МК, мембрана становится гораздо чувствительнее даже к небольшим колебаниям локальной концентрации калия. Эти небольшие вариации могут вызывать спонтанные ПД, еще больше усиливая миотонические симптомы. Этот эффект объясняет, почему блокада этих потенциалзависимых натриевых каналов является основной терапевтической целью при СК.
У здоровых людей высокая концентрация ионов калия нивелируется конкордантностью каналов CIC-1. Когда этот эффект утрачивается, как при МК, мембрана становится гораздо чувствительнее даже к небольшим колебаниям локальной концентрации калия. Эти небольшие вариации могут вызывать спонтанные ПД, еще больше усиливая миотонические симптомы. Этот эффект объясняет, почему блокада этих потенциалзависимых натриевых каналов является основной терапевтической целью при СК.
Эпидемиология
Частота СК исторически была описана как 1:23 000 для аутосомно-доминантной СК (болезнь Томсена) и 1:50 000 для аутосомно-рецессивной СК (болезнь Беккера). В настоящее время признано, что аутосомно-доминантная форма на самом деле встречается реже, чем аутосомно-рецессивная СК. Исследование 300 пациентов с СК, проведенное в Соединенном Королевстве, показало, что только 37% этой когорты обладали аутосомно-доминантными мутациями.[9] Оба типа СК чаще наблюдаются в Северной Скандинавии, с распространенностью 1 на 10 000. [10][11] Эта распространенность в десять раз превышает предполагаемую распространенность во всем мире.[12]
[10][11] Эта распространенность в десять раз превышает предполагаемую распространенность во всем мире.[12]
Анамнез и физикальное исследование
Основным признаком СК является замедленное расслабление сокращенных скелетных мышц. Признано, что существует значительная изменчивость фенотипа как аутосомно, так и рецессивно наследуемой СК; это может помешать быстрой диагностике и лечению. Идентичные мутации могут приводить к значительно различающимся клиническим проявлениям, а взаимосвязь между генотипом и фенотипом при этих каналопатиях считается чрезвычайно сложной.[13] И наоборот, некоторые специфические мутации, по-видимому, приводят к высоко воспроизводимому фенотипу. Подробный семейный анамнез часто полезен для выяснения аутосомно-рецессивного типа наследования, хотя при спорадических мутациях он может отсутствовать.
В раннем детстве симптомы могут включать трудности с кормлением, в том числе дисфагию, рефлюкс, рвотные позывы и удушье. Дети могут казаться неуклюжими и часто падать, даже после того, как они научились ходить. Пациенты также могут испытывать трудности с открытием глаз во время длительного сокращения, например, при плаче. Эти признаки могут быть незаметными; поэтому очень важны тщательный сбор анамнеза у пациентов и лиц, осуществляющих уход за ними, а также клиническое обследование. Симптомы часто сначала прогрессируют после того, как они впервые появились, а затем затухают. [14]
Пациенты также могут испытывать трудности с открытием глаз во время длительного сокращения, например, при плаче. Эти признаки могут быть незаметными; поэтому очень важны тщательный сбор анамнеза у пациентов и лиц, осуществляющих уход за ними, а также клиническое обследование. Симптомы часто сначала прогрессируют после того, как они впервые появились, а затем затухают. [14]
Ригидность мышц, наблюдаемая при СК, часто улучшается при выполнении упражнений или повторяющихся движений. Этот эффект называется феноменом «разогрева» (хотя он быстро теряется при прекращении активности). Мужчины, по-видимому, более тяжело страдают от СК, чем женщины. Кроме того, симптомы часто ухудшаются во время беременности и менструации; эти наблюдения подразумевают, что половые гормоны влияют на функцию канала CIC-1.
СК классически определяется как отдельные клинические заболевания, основанные на типе наследования. Между этими двумя заболеваниями есть тонкие различия. Учитывая обсуждавшуюся выше фенотипическую изменчивость, окончательный диагноз достигается с помощью генетического тестирования.
Болезнь Беккера (ББ): Это аутосомно-рецессивная наследуемая СК. Это связано с миотонией от умеренной до тяжелой и преходящей слабостью, хотя в некоторых случаях это может стать прогрессирующим. BD обычно проявляется позже в детстве, чем болезнь Томсена (TD) [17]. Мышечная гипертрофия более выражена при BD, чем при TD, и может быть особенно заметна в более крупных группах мышц нижних конечностей.
Болезнь Томсена (ТД): Доминантно наследуемый СК связан с более ранним началом симптомов, хотя они менее выражены, чем ББ. Легкая, прогрессирующая, постоянная слабость, описанная при BD, не связана с TD.[18] Аутосомно-доминантные мутации, вызывающие TD, связаны со сниженной пенетрантностью, а идентичные мутации, унаследованные из поколения в поколение, могут вызывать заметно различающиеся фенотипы.
Оценка
Клиническое обследование: Миотонию можно наблюдать, попросив пациента несколько раз открыть и закрыть глаза или разжать и сжать кулак. Повторное постукивание по мышце также вызывает миотонию. [19] Пациенты могут с трудом сразу разгибать пальцы после рукопожатия. Можно также продемонстрировать «эффект разогрева».
[19] Пациенты могут с трудом сразу разгибать пальцы после рукопожатия. Можно также продемонстрировать «эффект разогрева».
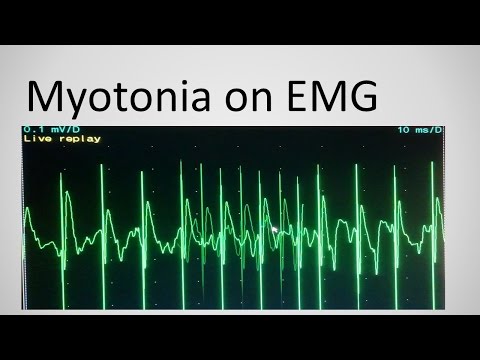
Биохимические исследования обычно ничем не примечательны, хотя было описано легкое повышение уровня креатининкиназы, которое в три-четыре раза превышало верхнюю границу нормы [19].] Электромиография является полезным инструментом для диагностики СК, однако она требует много времени, неудобна и приводит к перекрытию различных каналопатий.[20] Он может демонстрировать поток спонтанной электрической активности в виде диффузных миотонических разрядов или «всплесков». Электромиографической разницы между двумя типами MC нет. Учитывая широкую доступность генетического тестирования, биопсия мышц в настоящее время выполняется редко, но она может выявить гетерогенные мышечные волокна с увеличенным числом ядер и отсутствием волокон типа 2В [19].] Биопсия мышц не требуется для установления диагноза СК.
Генетическое тестирование считается золотым стандартом. Однако у многих пациентов мутации в гене CLCN1 не выявляются, несмотря на выраженные клинические признаки, коррелирующие с миотонической картиной. Сначала обычно выполняется мультигенная панель, включающая ген CLCN1, а также другие представляющие интерес гены, такие как SCN4A (обсуждается в дифференциальной диагностике). Генетическое тестирование и выбор используемых методов должны проводиться специализированными центрами для оценки вариантов неизвестной значимости (VUS), корреляции выявленных мутаций с клиническим фенотипом и минимизации затрат. Если мультигенная панель не идентифицирует объяснительную мутацию, могут быть использованы геномные методы, включая экзомное секвенирование и митохондриальное секвенирование.
Сначала обычно выполняется мультигенная панель, включающая ген CLCN1, а также другие представляющие интерес гены, такие как SCN4A (обсуждается в дифференциальной диагностике). Генетическое тестирование и выбор используемых методов должны проводиться специализированными центрами для оценки вариантов неизвестной значимости (VUS), корреляции выявленных мутаций с клиническим фенотипом и минимизации затрат. Если мультигенная панель не идентифицирует объяснительную мутацию, могут быть использованы геномные методы, включая экзомное секвенирование и митохондриальное секвенирование.
Лечение/управление
Фармакологическое лечение СК показано не всегда, и пациенты должны быть обследованы неврологом для оценки потребности в лекарствах до его начала.[21] Модификации образа жизни включают в себя избегание выявленных триггеров, таких как стресс и холод. Сообщается, что упражнения, особенно гимнастика, помогают уменьшить миотонию; однако эффект требует дальнейшего изучения.
Лекарства, которые используются, как правило, направлены на снижение гиперчувствительности мышечной мембраны путем блокирования потока ионов натрия. [22] Мексилетин является наиболее часто назначаемым лекарством, но требует ЭКГ-мониторинга интервала QT до и во время применения [23]. Он классифицируется как антиаритмический препарат класса 1b и является производным местного анестетика лидокаина. Мексилетин в основном используется для лечения желудочковых аритмий путем блокирования быстрого притока натрия, ответственного за фазу 0 потенциала действия (ПД), укорочения ПД и продления рефрактерного периода.[24]
[22] Мексилетин является наиболее часто назначаемым лекарством, но требует ЭКГ-мониторинга интервала QT до и во время применения [23]. Он классифицируется как антиаритмический препарат класса 1b и является производным местного анестетика лидокаина. Мексилетин в основном используется для лечения желудочковых аритмий путем блокирования быстрого притока натрия, ответственного за фазу 0 потенциала действия (ПД), укорочения ПД и продления рефрактерного периода.[24]
Побочные эффекты могут включать тремор, головокружение, атаксию и желудочно-кишечные расстройства. Эти побочные эффекты обычно зависят от дозы и обратимы при прекращении приема препарата или снижении дозы. Также широко используются фенитоин и другие противосудорожные препараты.[25]
Калиевые каналы — новая мишень. Ретигабин, активатор калиевых каналов, был исследован на мышиных моделях СК, и было показано, что он значительно снижает тяжесть миотонии in vivo [22]. Однако признано, что по-прежнему недостаточно доказательств, определяющих фармакологическое лечение СК.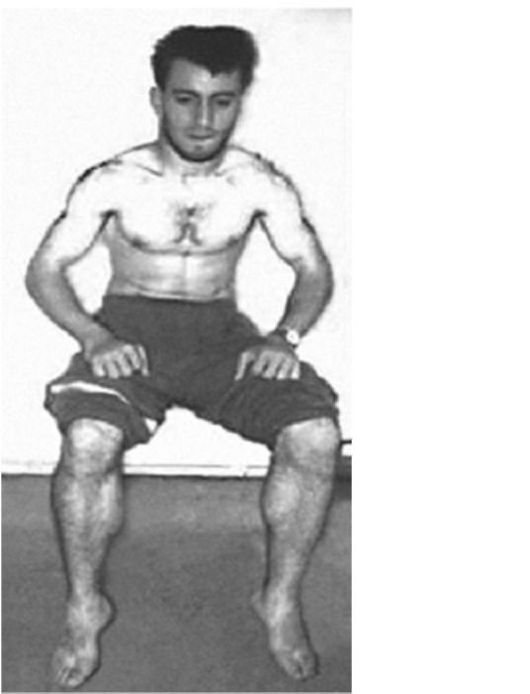 [26]
[26]
Дифференциальный диагноз
СК — наиболее распространенная недистрофическая миотония (НДМ). Состояние не является дистрофическим, потому что мышечная ткань не нарушена структурно как часть патологического процесса, а вместо этого миотония вызвана дефектными ионными каналами в скелетных мышцах, что приводит к нарушению ионной проводимости. В дополнение к дисфункциональным анионным каналам, наблюдаемым при СК, мутации в катионных каналах, а именно натриевых ионных каналах, вызывают врожденную парамиотонию (ВМК), калиевую миотонию (ПАМ) и калийчувствительный (гиперкалиемический) периодический паралич (ГиперПП) [3]. ]
Мутации в гене SCN4A приводят к потере функции этих потенциалзависимых натриевых каналов. Симптомы обычно хуже в нижних конечностях при СК (пациенты часто с трудом могут быстро встать), тогда как при ЛАМ более сильно поражаются глаза и лицо. Миотония, связанная с HyperPP, легкая и особенно связана с веками и языком.[19]
Отличить СК от других заболеваний часто возможно клинически при тщательном учете усугубляющих или облегчающих факторов, наличии внемышечного заболевания и данных электромиографии. Симптомы и особенности нарушений калиевых каналов ухудшаются после приема пищи, богатой калием, и пациенты могут описывать свою миотонию как болезненную. Эта история не типична для МС. Миотонические дистрофии также являются важным дифференциальным признаком, который следует учитывать при исследовании мышечной слабости и спастичности в раннем детстве и младенчестве. Миотоническая дистрофия (МД) I и II типа связана с системными проявлениями, включая эндокринную дисфункцию, дефекты сердечной проводимости и образование катаракты. Кроме того, картина мышечной слабости, наблюдаемая при MD, сильно отличается от наблюдаемой при MC.
Симптомы и особенности нарушений калиевых каналов ухудшаются после приема пищи, богатой калием, и пациенты могут описывать свою миотонию как болезненную. Эта история не типична для МС. Миотонические дистрофии также являются важным дифференциальным признаком, который следует учитывать при исследовании мышечной слабости и спастичности в раннем детстве и младенчестве. Миотоническая дистрофия (МД) I и II типа связана с системными проявлениями, включая эндокринную дисфункцию, дефекты сердечной проводимости и образование катаракты. Кроме того, картина мышечной слабости, наблюдаемая при MD, сильно отличается от наблюдаемой при MC.
Прогноз
Ни аутосомно-, ни рецессивно наследуемые СК не связаны с системными эффектами и не ограничивают ожидаемую продолжительность жизни.[21] Этот прогноз существенно отличается от течения миотонической дистрофии, поэтому очень важна точная диагностика. После появления симптомов СК эта группа заболеваний обычно не прогрессирует. Обычно считается, что болезнь Беккера связана с более выраженными симптомами, чем при болезни Томсена, и со временем может сохраняться постоянная слабость. Пациенты с СК должны иметь доступ к услугам генетического консультирования, чтобы принимать информированные решения относительно планирования семьи.
Пациенты с СК должны иметь доступ к услугам генетического консультирования, чтобы принимать информированные решения относительно планирования семьи.
Осложнения
Необходимо соблюдать осторожность во время анестезии, особенно с деполяризующими миорелаксантами, которые связаны с побочными эффектами у пациентов с СК, такими как глубокий мышечный спазм и трудности при вторичной вентиляции [27]. Лекарства, которых следует избегать или, по крайней мере, использовать с осторожностью у пациентов с СК, включают:
Суксаметоний – может вызывать глубокий мышечный спазм и трудности с вентиляцией.
Адреналин и бета-агонисты — могут вызывать ухудшение симптомов
Бета-антагонист — потенциально может усиливать тяжесть симптомов.
Колхицин – может вызвать миопатию, особенно у лиц с почечной недостаточностью.
Беременность связана с ухудшением СК; поэтому межпрофессиональная команда необходима на протяжении всей беременности, родов и послеродового периода.
Сдерживание и обучение пациентов
Обучение и поддержка пациентов с СК и их семей имеют важное значение. Физические проявления миотонии и ее последствия могут сильно изнурять и оказывать глубокое психологическое воздействие на больных пациентов.[20] И наоборот, из-за мышечной гипертрофии, связанной с СК, пациенты сообщают, что они подвергаются преследованиям и дискриминации, поскольку кажутся здоровыми. Пациенты могут пытаться «бороться» с миотонией, которая может ухудшить эффект, а стресс, как известно, усугубляет симптомы. Поэтому жизненно важны стратегии выживания, направленные на повышение функциональности, с уделением особого внимания психологическому здоровью. Может потребоваться корректировка домашней, школьной и рабочей среды, чтобы снизить риск падений и уменьшить их последствия, когда они происходят. Для повышения безопасного глотания и снижения вероятности аспирации может потребоваться изменение диеты.
Улучшение результатов работы команды здравоохранения
СК — это очень изменчивое, но потенциально разрушительное состояние, поражающее пациентов и их семьи с раннего детства на протяжении всей жизни. Поскольку это генетическое заболевание, генетическое тестирование и консультирование играют важную роль, чтобы проверить других членов семьи и дать рекомендации по планированию семьи. Таким образом, эффективное и безопасное лечение СК требует координации нескольких медицинских специальностей, включая педиатрию и клиническую генетику, а также участия физиотерапевтов, диетологов, эрготерапевтов и психологов. Роль этой межпрофессиональной команды теперь хорошо известна в специализированных центрах.
Поскольку это генетическое заболевание, генетическое тестирование и консультирование играют важную роль, чтобы проверить других членов семьи и дать рекомендации по планированию семьи. Таким образом, эффективное и безопасное лечение СК требует координации нескольких медицинских специальностей, включая педиатрию и клиническую генетику, а также участия физиотерапевтов, диетологов, эрготерапевтов и психологов. Роль этой межпрофессиональной команды теперь хорошо известна в специализированных центрах.
Контрольные вопросы
Доступ к бесплатным вопросам с несколькими вариантами ответов по этой теме.
Комментарий к этой статье.
Ссылки
- 1.
Лоссин С., Джордж А.Л. Врожденная миотония. Ад Генет. 2008;63:25-55. [PubMed: 19185184]
- 2.
Conravey A, Santana-Gould L. Врожденная миотония и миотоническая дистрофия: наблюдение и лечение. Варианты лечения Curr Neurol. 2010 Январь; 12(1):16-28.
[В паблике: 20842486]- 3.
Platt D, Griggs R. Каналопатии скелетных мышц: новый взгляд на периодические параличи и недистрофические миотонии. Карр Опин Нейрол. 2009 окт; 22 (5): 524-31. [PMC free article: PMC2763141] [PubMed: 19571750]
- 4.
Sun C, Tranebjaerg L, Torbergsen T, Holmgren G, Vanhhelue M. Spectrum of Clcn1 Mattions у пациентов с Myotnia in Scandita in. Eur J Hum Genet. 2001 г., декабрь 9(12):903-9. [В паблике: 11840191]
- 5.
Li L, McCall C, Hu X. Редакционная статья: Программирование врожденного иммунитета и память при разрешении и неразрешении воспаления. Фронт Иммунол. 2020;11:177. [Бесплатная статья PMC: PMC7026667] [PubMed: 32117304]
- 6.
Zhang J, Bendahhou S, Sanguinetti MC, Ptácek LJ. Функциональные последствия мутаций гена хлоридного канала (CLCN1), вызывающих врожденную миотонию. Неврология. 2000 22 февраля; 54 (4): 937-42. [PubMed: 10690989]
- 7.
Тан CY, Чен TY. Физиология и патофизиология ХЛК-1: механизмы заболевания хлоридных каналов, миотония. Дж. Биомед Биотехнолог. 2011;2011:685328. [Бесплатная статья PMC: PMC3237021] [PubMed: 22187529]
- 8.
Lipicky RJ, Bryant SH, Salmon JH. Кабельные параметры, содержание натрия, калия, хлоридов и воды, а также отток калия в изолированных наружных межреберных мышцах здоровых добровольцев и пациентов с врожденной миотонией. Джей Клин Инвест. 1971 Октябрь; 50 (10): 2091-103. [Бесплатная статья PMC: PMC292143] [PubMed: 4940295]
- 9.
Фиальо Д., Шорге С., Пуковска У., Дэвис Н.П., Лабрум Р., Хаворт А., Стэнли Э., Суд Р., Уэйклинг В., Дэвис М.Б., Кульманн Д.М., Ханна М.Г. Миотония хлоридных каналов: горячая точка экзона 8 для доминантно-негативных взаимодействий. Мозг. 2007 г., декабрь; 130 (часть 12): 3265-74. [PubMed: 17932099]
- 10.
Coote DJ, Davis MR, Cabrera M, Needham M, Laing NG, Nowak KJ.
Карта клинических полезных генов для: врожденной миотонии по аутосомно-доминантному типу (болезнь Томсена). Eur J Hum Genet. 2018 июль; 26 (7): 1072-1077. [Бесплатная статья PMC: PMC6018704] [PubMed: 29695755]- 11.
Papponen H, Toppinen T, Baumann P, Myllylä V, Leisti J, Kuivaniemi H, Tromp G, Myllylä R. Мутации основателя и высокая распространенность врожденной миотонии в северной Финляндии. Неврология. 1999 г., 22 июля; 53 (2): 297–302. [PubMed: 10430417]
- 12.
Эмери А.Э. Популяционные частоты наследственных нервно-мышечных заболеваний — всемирный обзор. Нервно-мышечное расстройство. 1991;1(1):19-29. [PubMed: 1822774]
- 13.
Моралес Ф., Пуш М. Современный обзор сложности отношений генотип-фенотип при миотонических каналопатиях. Фронт Нейрол. 2019;10:1404. [Бесплатная статья PMC: PMC6978732] [PubMed: 32010054]
- 14.
Kuhn E. [Врожденная миотония (Томсен) и рецессивная генерализованная миотония (Беккер)].
Нервенарцт. 1993 декабрь; 64 (12): 766-9. [PubMed: 8114977]- 15.
Колдинг-Йоргенсен Э. Фенотипическая изменчивость при врожденной миотонии. Мышечный нерв. 2005 июль; 32 (1): 19-34. [PubMed: 15786415]
- 16.
Fialho D, Kullmann DM, Hanna MG, Schorge S. Негеномные эффекты половых гормонов на CLC-1 могут способствовать гендерным различиям при врожденной миотонии. Нервно-мышечное расстройство. 2008 ноябрь; 18 (11): 869-72. [PubMed: 18815035]
- 17.
Дуно М., Колдинг-Йоргенсен Э., Груннет М., Йесперсен Т., Виссинг Дж., Шварц М. Различия в аллельной экспрессии гена CLCN1 и возможное влияние на фенотип миотонии врожденного тип . Eur J Hum Genet. 2004 Сентябрь; 12 (9)):738-43. [PubMed: 15162127]
- 18.
Ричардсон RC, Tarleton JC, Bird TD, Gospe SM. Усечение мутаций CLCN1 при врожденной миотонии: различные модели наследования. Мышечный нерв. 2014 Апрель; 49 (4): 593-600. [PubMed: 23893571]
- 19.
Hahn C, Salajegheh MK. Миотонические расстройства: обзорная статья. Иран Дж Нейрол. 2016 05 января; 15 (1): 46-53. [Бесплатная статья PMC: PMC4852070] [PubMed: 27141276]
- 20.
Phillips L, Trivedi JR. Каналопатии скелетных мышц. Нейротерапия. 2018 Окт;15(4):954-965. [Бесплатная статья PMC: PMC6277285] [PubMed: 30341599]
- 21.
Gutmann L, Phillips LH. Врожденная миотония. Семин Нейрол. 1991 Сентябрь; 11 (3): 244-8. [PubMed: 1947487]
- 22.
Dupont C, Denman KS, Hawash AA, Voss AA, Rich MM. Лечение врожденной миотонии ретигабином у мышей. Опыт Нейрол. 2019 Май; 315:52-59. [Бесплатная статья PMC: PMC6431423] [PubMed: 30738808]
- 23.
Roberts K, Kentris M. StatPearls [Интернет]. Издательство StatPearls; Остров сокровищ (Флорида): 8 мая 2022 г. Миотония. [В паблике: 32644698]
- 24.
Сингх С., Керндт С.С., Чаухан С., Зельцер Р. StatPearls [Интернет].
 [В паблике: 20842486]
[В паблике: 20842486]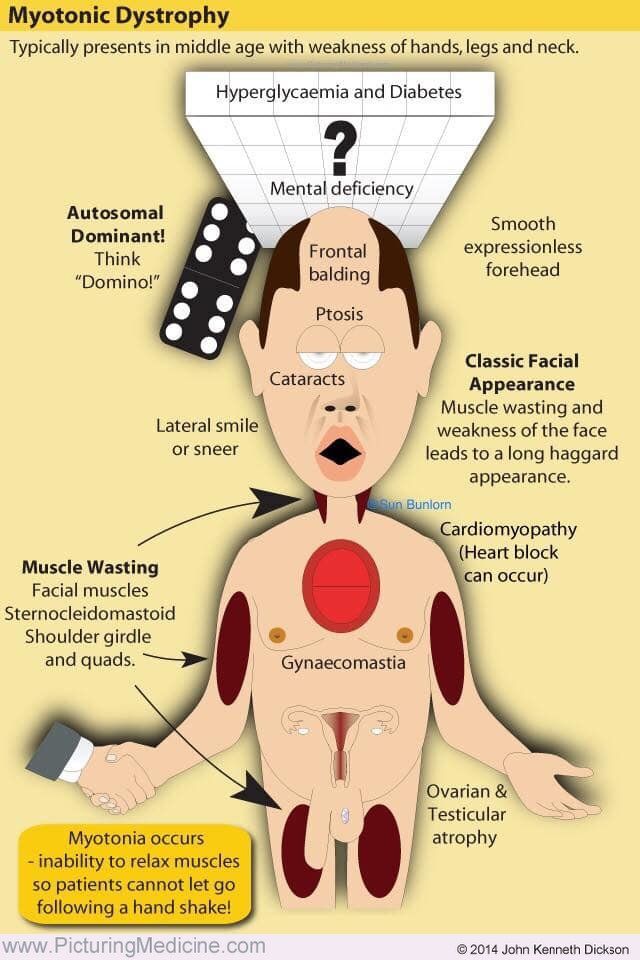
 Карта клинических полезных генов для: врожденной миотонии по аутосомно-доминантному типу (болезнь Томсена). Eur J Hum Genet. 2018 июль; 26 (7): 1072-1077. [Бесплатная статья PMC: PMC6018704] [PubMed: 29695755]
Карта клинических полезных генов для: врожденной миотонии по аутосомно-доминантному типу (болезнь Томсена). Eur J Hum Genet. 2018 июль; 26 (7): 1072-1077. [Бесплатная статья PMC: PMC6018704] [PubMed: 29695755] Нервенарцт. 1993 декабрь; 64 (12): 766-9. [PubMed: 8114977]
Нервенарцт. 1993 декабрь; 64 (12): 766-9. [PubMed: 8114977]
