
Мышечная дистрофия это: Мышечная дистрофия | Неврология | Заболевания
Мышечная дистрофия | Неврология | Заболевания
Мышечная дистрофия – это патологическое заболевание, которое характерно для людей, ведущих лежачий образ жизни. Также данное заболевание может появится у людей с острыми хроническими заболеваниями мышц и костей.
Виды
Мышечная дистрофия очень распространенное патологическое заболевание. Бывает детская и взрослая дистрофия мышц. Также мышечная дистрофия имеет наследственный характер (генетическая и наследственная дистрофия). По характеру и месту локализации различают:
-
инфекционную и неинфекционную;
-
миотоническую;
-
тазово-плечевую;
-
врожденную;
-
плечелопаточную;
.
Симптомы
Мышечная дистрофия прогрессирует заболевание мышечной слабости и потери трудоспособности
Диагностика
Обратиться к доктору следует немедленно, как только вы заметили мышечную слабость.. К методам диагностики на данном этапе развития медицины относят МРТ. Оно покажет анатомические и физиологические изменения в организме. При этом заболевании также стоит сдать общий анализ крови, мочи и кала. После проведенных диагностик доктор поставит диагноз и направит на лечение.
Лечение
Лечение проводиться с помощью комплексной терапии: консервативное лечение и физиотерапия. Еще не разработано лечение, которые бы полностью устранило это заболевание.
Профилактика
Лечение мышечной дистрофии продолжается его профилактикой. Очень важно после выписки с госпиталя не забыть о приписках врача. Пациент должен снизить к минимуму физические нагрузки, стрессовые ситуации, вести здоровый образ жизни, отказаться от алкоголя, наркотиков и курения.
Прогрессирующие мышечные дистрофии | МКДЦ ФГБНУ НЦН
Прогрессирующие мышечные дистрофии (ПМД) — гетерогенная группа наследственных заболеваний, характеризующихся прогрессирующей мышечной слабостью и атрофией скелетных мышц.
КЛИНИЧЕСКАЯ КАРТИНА
Для всех ПМД типичны мышечная слабость различной степени выраженности и мышечные атрофии. Тип распределения мышечной слабости при ПМД — один из основных диагностических критериев. Для каждой из форм ПМД характерно избирательное поражение определённых мышц при сохранности других, рядом расположенных. В целом типичный миопатический симптомокомплекс включает следующие признаки.
• Симметричную проксимальную мышечную слабость различной степени выраженности (мышечная сила от 3-4 баллов на ранней и до 1-0 — на поздних стадиях заболевания) , постепенно развивающиеся атрофии мышц.
• Симптом Говерса: больной, для того чтобы подняться из положения на корточках, опирается руками об пол, затем поднимается, опираясь руками об колени, — «взбирается по себе». Этот рано появляющийся симптом обусловлен слабостью мышц бёдер и тазового пояса.
• Затруднения при ходьбе по лестнице — больной помогает себе с помощью рук.
• «Утиную» (переваливающуюся) походку, связанную со слабостью мышц тазового пояса.
• Поясничный гиперлордоз, обусловленный слабостью мышц тазового пояса и спины.
• «Крыловидные» лопатки вследствие слабости передней зубчатой мышцы, а также других мышц, фиксирующих лопатку.
• Псевдогипертрофию икроножных мышц вследствие развития в них соединительной ткани (сила мышц при этом снижена) .
• Ходьбу на цыпочках из-за контрактур ахилловых сухожилий.
• Сохранность экстраокулярных мышц, мышц лица.
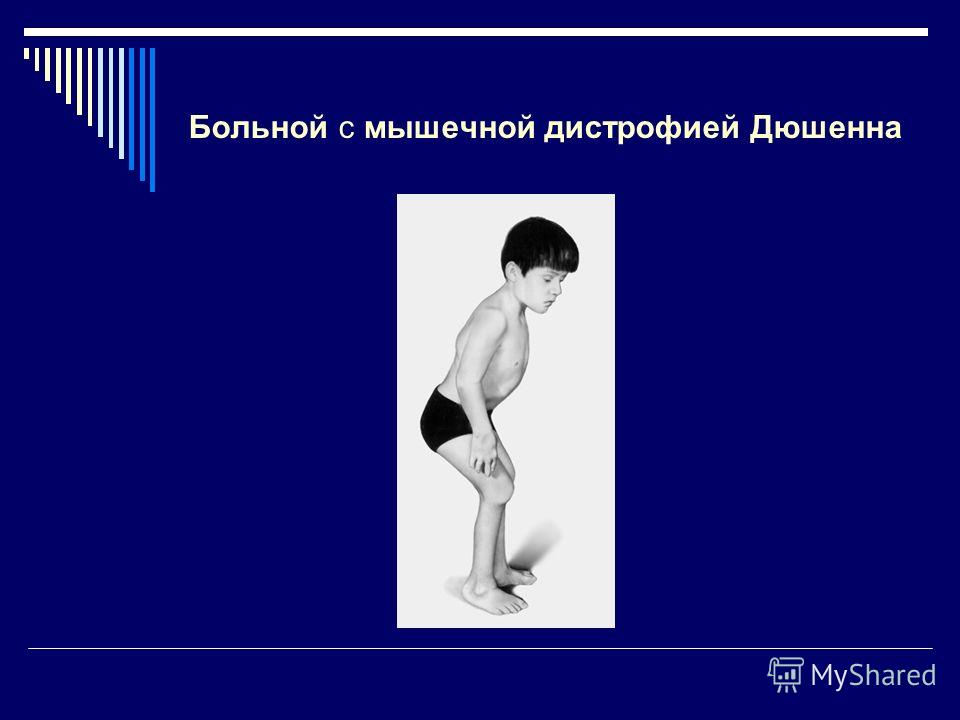
Миопатический симптомокоплекс наиболее отчётливо выявляют при ПМД Дюшенна и Беккера.
• Для ПМД Дюшенна характерно раннее начало заболевания (в 3-7 лет) , быстрое прогрессирование, высокие показатели КФК, выраженная спонтанная активность по данным игольчатой ЭМГ, отсутствие дистрофина в мышцах при иммуногистохимическом исследовании. По мере прогрессирования мышечной слабости затрудняется самостоятельная ходьба и уже в 9-15 лет больные вынуждены пользоваться инвалидным креслом, что провоцирует развитие кифосколиоза, остеопороза. На поздних стадиях у большинства больных развиваются дилатационная кардиомиопатия, слабость дыхательной мускулатуры. Интеллект чаще всего умеренно снижен.
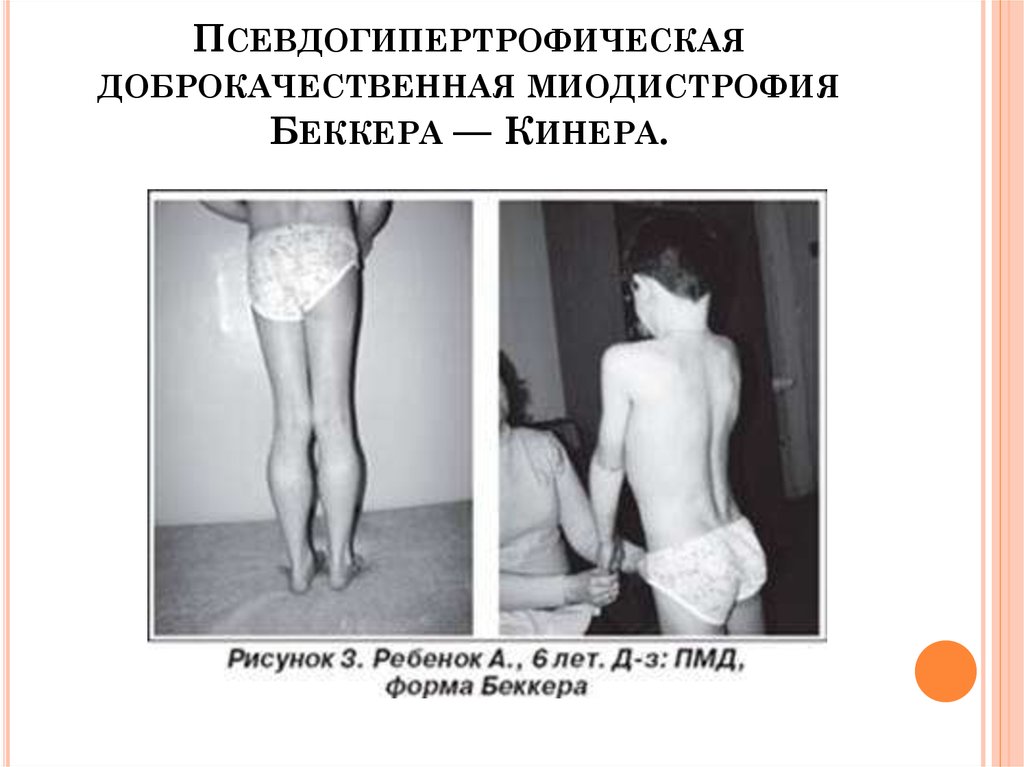
• Клинические про явления ПМД Беккера в целом напоминают таковые при форме Дюшенна, но течение заболевания более мягкое: дебют приходится на более поздний возраст (от 2 до 21 года, в среднем в 11 лет) , летальный исход наступает позже (в 23-63 года) .
• Конечностно-поясные формы ПМД также характеризуются развитием миопатического симптомокомплекса. ПМД Эрба по возрасту начала заболевания, скорости прогрессирования и клиническим проявлениям напоминает форму Беккера, однако для неё не характерна кардиальная патология, кроме того, заболевание отмечают как у мальчиков, так и девочек. При других конечностнопоясных формах возможны слабость мышц лица и кардиомиопатия.
• ПМД Ландузи-Дежерина характеризуется выраженной слабостью мимических мышц (за исключением редкой формы без мимической слабости), симптомом «крыловидных» лопаток, слабостью дву- и трёхглавых мышц плеча при интактных дельтовидных мышцах, степпажем. Как правило, интактными остаются экстраокулярные мышцы (за исключением одного подтипа) и мышцы языка и глотки, дыхательная мускулатура. У некоторых больных возникает слабость мышц тазового пояса (около 20% больных вынуждены пользоваться инвалидным креслом) . Мышечные атрофии часто бывают асимметричными. У многих больных отмечают снижение слуха, кардиомиопатию или нарушения сердечного ритма.
У многих больных отмечают снижение слуха, кардиомиопатию или нарушения сердечного ритма.
• ПМД Эмери-Дрейфуса характеризуется наличием контрактур (чаще в локтевых, коленных суставах, задних мышцах шеи, из-за которых голова оказывается слегка запрокинутой) и плечелопаточно-перонеальным распределением мышечной слабости и атрофий с сохранностью лицевой мускулатуры. Часто отмечают нарушения ритма сердца и кардиомиопатию. Заболевание часто дебютирует с контрактур.
• Основной симптом офтальмофарингеальной формы — хроническая прогрессирующая наружная офтальмоплегия, затем присоединяется умеренный бульбарный синдром. В дальнейшем развивается проксимальная мышечная слабость в руках и ногах.
• Дистальные миопатии характеризуются преобладанием слабости дистальных мышц. При миопатии Веландер в наибольшей степени поражаются разгибатели кистей, при миопатии Миоши — икроножные мышцы: больные плохо стоят на носках, часто спотыкаются. При миопатии Говерса, тибиальной миопатии главный симптом — степпаж из-за слабости перонеальной группы мышц, при этом миопатия Говерса склонна к дальнейшей генерализации: через 5-10 лет присоединяется слабость кистей и мышц шеи, часто отмечают «свисание» 1 пальца на ногах и V — на руках. При тибиальной миопатии, распространённой в Финляндии, чаще всего наблюдают изолированное поражение передних больше берцовых мышц, иногда развивается кардиомиопатия.
СИМПТОМЫ
При ПМД Дюшенна, Беккера, конечностно-поясных формах проявляется наиболее выраженная слабость в пояснично-подвздошных мышцах, мышцах бёдер, дельтовидных, дву- и трёхглавых мышцах плеча. Менее выражена слабость в дистальных мышцах конечностей. Лицевые мышцы остаются сохранными. Наряду с мышечной слабостью постепенно развиваются гипотрофии поражённых мышц вплоть до атрофии на поздних стадиях. При этом соседние мышцы могут быть полностью клинически интактны.
ЛЕЧЕНИЕ
Лечение должно производиться исключительно врачом-неврологом. Самолечение недопустимо. В настоящий момент радикального лечения ПМД не существует. Цель лечения — поддержание мышечной силы, предупреждение развития контрактур, деформаций суставов.
В настоящий момент радикального лечения ПМД не существует. Цель лечения — поддержание мышечной силы, предупреждение развития контрактур, деформаций суставов.
Немедикаментозное лечение
Чрезмерная физическая нагрузка, как и недостаточная, приводит к нарастанию мышечной слабости. Ежедневная ЛФК позволяет поддерживать мышечный тонус и препятствует развитию контрактур. Комплекс ЛФК обязательно должен включать активные и пассивные упражнения, упражнения на растяжку/предупреждение контрактур и дыхательную гимнастику. Активный массаж с разминанием мышц может усиливать мышечную слабость и утомляемость, поэтому рекомендуют щадящий массаж. Физиотерапевтическое лечение больные переносят по-разному: некоторые не ощущают улучшений или даже жалуются на усиление мышечной слабости.
Хирургическое лечение
В некоторых случаях возможно хирургическое лечение контрактур, однако при этом необходимо помнить о возможности увеличения мышечной слабости за время восстановительного лечения (вплоть до потери способности к ходьбе). В ряде случаев необходима имплантация кардиостимулятора.
Мышечная дистрофия: Формы,Причины,Диагностика | Doc.ua
В результате развития этого заболевания человек становится инвалидом. Позвоночник деформируется, теряется двигательная способность. Заболевание затрагивает и другие системы организма – развивается сердечная и дыхательная недостаточность, происходят нарушения интеллекта, ухудшается память и теряется способность к обучению.
Формы
Различают несколько форм заболевания.
Мышечная дистрофия Дюшена чаще всего проявляется уже в детском возрасте. Болеют ею в большинстве своем мальчики. Первые симптомы проявляются уже в возрасте 2-5 лет. Изначально слабость охватывает группы мышц ног и тазового пояса. Затем болезнь поражает все тело.
Дегенерация мышечной ткани приводит к увеличению в объеме икроножных мышц, жировая и соединительная ткани быстро разрастаются.
Заболевание прогрессирует довольно стремительно: уже в 12 лет больные не способны передвигаться, а к 20-ти годам большинство из них умирает.
Миодистрофия Беккера встречается редко, и страдают ею преимущественно низкорослые люди. В отличие от предыдущей формы, имеет не такое стремительное развитие. Достаточно долго человек пребывает в удовлетворительном состоянии. Если диагностирована прогрессирующая мышечная дистрофия, как еще называют эту форму заболевания, то инвалидизация случается только на фоне сопутствующих травм и заболеваний.
Мышечная дистрофия Эрба развивается в подростковом возрасте – 10-20 лет. В этом случае мышцы поражаются в направлении сверху вниз: сначала разрушительный процесс затрагивает мышцы плечевого пояса, затем таза, нижних конечностей. Больного можно определить по характерной походке и осанке: он переваливается, живот выпячен вперед, грудина отодвинута назад. Прогрессирует эта форма болезни медленно.
Миотоническая форма дистрофии мышц чаще поражает взрослых – мужчин и женщин в возрасте от 20-40 лет, но в некоторых случаях она встречается и у младенцев. Это состояние характеризуется замедленным расслаблением мышц после сокращения (миотония), ослаблением мимических мышц. Особенность этой формы патологии состоит в том, что кроме скелетных поражаются и внутренние мышцы, в том числе сердечная. Деструктивный процесс может затрагивать и другие группы мышц, включая руки и ноги. Протекает заболевание вяло.
Плече-лопаточно-лицевая дистрофия мышц Ландузи-Дежерина поражает людей разного возраста – от 6 до 52 лет. Основная группа риска — подростки 10-15 лет. Сначала происходит дегенерация лицевых мышц, затем – туловища и конечностей. Первые симптомы – неполное смыкание век, губ, что ухудшает дикцию. Пациент длительное время сохраняет трудоспособность. Однако через 15-25 лет атрофируются мышцы тазового пояса. Это ухудшает двигательную функцию.
Причины
Пока досконально не изучены все механизмы развития дистрофии мышечных тканей у человека. Основной причиной называют мутацию гена, отвечающего за способность клеток мышц синтезировать специальные белки.
На сегодняшний день обнаружен ген, вызывающий патологию Дюшена. Он расположен в Х-хромосоме. Мать с дефектным геном передает его своему сыну, который заболевает, достигая 2-5-летнего возраста.
Он расположен в Х-хромосоме. Мать с дефектным геном передает его своему сыну, который заболевает, достигая 2-5-летнего возраста.
У женщин причиной развития патологии является и изменение гормонального фона, произошедшее по причине беременности, климакса или связанное с началом менструации.
Симптомы
Первые симптомы мышечной дистрофии некоторых форм заболевания могут появляться в самом раннем возрасте – от двух лет. Ребенок часто падает, быстро устает даже при небольших физических нагрузках, он неуклюж в движениях, что становится особенно заметно на фоне ровесников.
Мышечная дистрофия у детей до года проявляется в резкой деградации развития в сравнении с изначально нормальным ростом и жизнедеятельностью. Малыш утрачивает полученные ранее навыки – теряет способность держать головку, перестает самостоятельно переворачиваться. Как правило, такие дети не доживают до 3-х лет.
О развитии данного заболевания у взрослых говорит снижение мышечного тонуса, из-за слабости нарушается походка, атрофируются скелетные мышцы. Пациент не ощущает мышечной боли при стабильной чувствительности. Он жалуется на постоянную усталость. Происходит разрастание и увеличение икроножных мышц.
Диагностика
Врач невролог собирает семейный анамнез, стараясь выяснить, была ли у кого-нибудь из родственников подобная патология, выясняет характер дистрофии. Эти данные помогут составить дальнейший прогноз для пациента.
Проводится электромиография, во время которой исследуется электрическая активность мышечной ткани, при которой обнаруживается первичная мышечная дистрофия, отбирается образец ткани для изучения дистрофических изменений ткани и жировых отложений. При наличии современного оборудования также проводится молекулярно-биологический и иммунологический анализ для определения вероятности развития заболевания детей.
Лечение
Миодистрофия – неизлечимое заболевание. Все мероприятия терапии направлены на замедление разрушительных процессов в мышцах. Для этого пациенту назначают витамины группы В, кортикостероиды, аденозинтрифосфат.
Для этого пациенту назначают витамины группы В, кортикостероиды, аденозинтрифосфат.
Затормозить или даже остановить процесс дегенерации помогают препараты на основе фетальных стволовых клеток.
При лечении мышечной дистрофии эффективна физиотерапия. Она направлена на минимизацию развития контрактур и деформаций, для чего разрабатываются комплексы специальных физических упражнений.
Свою действенность доказал лечебный массаж. Если существует риск поражения дыхательной мускулатуры, проводится дыхательная гимнастика.
Профилактика
Перед планированием беременности женщине необходимо пройти обследование на предмет наличия патологических генов, особенно, если кто-нибудь из родственников имел такое заболевание. Выявить его можно даже у плода. Для этого изучается амниотическая жидкость, клетки плода или его кровь.
Мышечная дистрофия, описание заболевания на портале Medihost.ru
Причины мышечной дистрофии
Мышечная дистрофия – это заболевание мышц, точнее, группа заболеваний, которая приводит к слабости и дегенеративным изменениям в мышцах. Дистрофия чаще затрагивает скелетные мышцы. Они постепенно слабеют, теряют способность сокращаться, уменьшаются в объеме и замещаются соединительной или жировой тканью.
Причина заболевания – мутация гена, который отвечает за синтез белков в клетках мышечной ткани. Поражению могут подвергнуться различные виды генов – как расположенные в половой хромосоме, так и не связанные с ней. От этого зависит механизм передачи заболевания по наследству (например, ген Дюшенна, связанный с половой Х-хромосомой, передается по мужской линии; женщины не болеют дистрофией, вызываемой этим геном, хотя и являются его носителями).
Формы и симптомы мышечной дистрофии
Формы:
- Мышечная дистрофия Дюшенна – ею страдают, в основном, мальчики; проявляется в возрасте 2-5 лет, сначала поражаются мышцы ног и области таза, затем – более высоко расположенные мышцы. Из-за распада мышечной ткани и разрастания жировой и соединительной икроножные мышцы увеличиваются в объеме.
 Заболевание быстро прогрессирует, из-за чего к 12 годам человек уже не может двигаться, а к 20 годам большинство больных погибает.
Заболевание быстро прогрессирует, из-за чего к 12 годам человек уже не может двигаться, а к 20 годам большинство больных погибает. - Миотоническая мышечная дистрофия – чаще всего развивается у мужчин и женщин от 20 до 40 лет, медленно прогрессирует. Ее отличают слабость мышц лица и замедленное расслабление мышц после их сокращения. Поражаются скелетные мышцы, мышцы лица, конечностей, может поражаться и сердечная мышца, и мышцы внутренних органов.
- Мышечная дистрофия Беккера – редко встречающаяся форма, которая развивается достаточно медленно и позволяет людям, страдающим ею, в течение долгого времени сохранять удовлетворительное состояние. Осложнения возникают только из-за сопутствующих травм и заболеваний. Поражает чаще всего людей невысокого роста.
- Юношеская форма Эрба-Рота – заболевание развивается в возрасте 10-20 лет и начинается с атрофии мышц рук, плечевого пояса, затем – тазовой области и ног. Медленно прогрессирует.
- Плече-лопаточно-лицевая форма мышечной дистрофии – начинается с поражения лицевых мышц, затем – постепенной атрофии мышц плечевого пояса, туловища, рук и ног. Проявляется на ранних стадиях недостаточным смыканием век, губ, проблемами с дикцией. Трудоспособность сохраняется на протяжении долгого времени, проблемы начинаются только спустя 15-25 лет после начала заболевания, когда из-за атрофии тазовых мышц больному становится трудно двигаться.
 Заболевание быстро прогрессирует, из-за чего к 12 годам человек уже не может двигаться, а к 20 годам большинство больных погибает.
Заболевание быстро прогрессирует, из-за чего к 12 годам человек уже не может двигаться, а к 20 годам большинство больных погибает.Симптомами, общими для всех форм, являются:
- снижение мышечного тонуса;
- нарушения походки, частые падения;
- постепенная утрата физических навыков (у детей) – таких, как умение сидеть, ходить и пр.;
- отсутствие мышечных болей и нормальная чувствительность при выраженной слабости мышц, постоянные жалобы на усталость;
- атрофия скелетных мышц;
- увеличение мышц в размерах (особенно икр) из-за замещения мышечной ткани жировой и соединительной.
Для диагностики врач исследует семейный анамнез, чтобы выяснить наличие случаев заболевания в семье. Исследования, которые проводятся на этапе диагностики – электромиография, микроскопическое исследование участка ткани мышц, при наличии технической возможности – иммунологические и молекулярно-биологические исследования.
Исследования, которые проводятся на этапе диагностики – электромиография, микроскопическое исследование участка ткани мышц, при наличии технической возможности – иммунологические и молекулярно-биологические исследования.
Лечение мышечной дистрофии
Излечение мышечной дистрофии невозможно. Терапия в состоянии лишь замедлить дистрофию мышц с помощью введения медикаментов, содержащих витамин В1, кортикостероиды, стволовые клетки, аденозинтрифосфат и др. Также проводится физиолечение, лечебная гимнастика, дыхательная гимнастика.
Современные методики позволяют предсказать риск развития мышечной дистрофии у ребенка еще во время беременности.
МЫШЕЧНАЯ ДИСТРОФИЯ | Энциклопедия Кругосвет
Содержание статьиМЫШЕЧНАЯ ДИСТРОФИЯ, группа хронических наследственных заболеваний скелетных (произвольных) мышц человека. Мышечные дистрофии проявляются прогрессирующей слабостью и дегенерацией мышц.
Мышечная дистрофия Дюшенна
(псевдогипертрофическая мышечная дистрофия) – самая частая форма этого заболевания у детей. Причина болезни – генетический дефект, локализованный на X-хромосоме (одной из двух хромосом, определяющих пол человека). Женщины с дефектным геном передают его своим детям, но у них самих симптомы дистрофии, как правило, отсутствуют. У мальчиков, получивших дефектный ген, в возрасте от двух до пяти лет неизбежно развивается мышечная слабость.
В первую очередь страдают крупные мышцы нижних конечностей и тазового пояса. Затем дегенерация распространяется на мышцы верхней половины тела, а далее постепенно и на все основные группы мышц.
Мышечная дистрофия Дюшенна – одна из самых тяжелых и быстро прогрессирующих форм. К 12 годам больные обычно теряют способность передвигаться, а к 20 годам большинство из них погибает.
Миотоническая мышечная дистрофия
(болезнь Штейнерта) – наиболее распространенная форма мышечной дистрофии у взрослых. Она обусловлена дефектным геном на 19-й хромосоме. Мужчины и женщины страдают в равной степени и могут передать генетический дефект детям. Заболевание проявляется в любом возрасте, в том числе в младенчестве, но чаще всего – между 20 и 40 годами. Первыми симптомами служат миотония (замедленное расслабление мышц после сокращения) и слабость мимических мышц; возможно также поражение мышц конечностей и других частей тела. Прогрессирование болезни происходит в большинстве случаев медленно, и полная инвалидность может наступить не ранее чем через 15 лет.
Особенность данного заболевания состоит в том, что помимо произвольных мышц оно поражает также гладкую мускулатуру и сердечную мышцу.
Патоморфология.
Все формы мышечной дистрофии характеризуются дегенерацией мышц, но не связанных с ними нервов. В пораженной мышечной ткани обнаруживают различные изменения, в том числе значительные колебания толщины (диаметра) мышечных волокон. Постепенно эти волокна теряют способность сокращаться, распадаются и замещаются жировой и соединительной тканью.
Диагноз.
По своим клиническим проявлениям мышечные дистрофии сходны со спинальными амиотрофиями – наследственными болезнями, поражающими двигательные нейроны спинного мозга. Эти болезни тоже приводят к выраженной мышечной слабости, иногда угрожающей жизни. Для подтверждения диагноза мышечной дистрофии может потребоваться электромиография, а иногда и биопсия мышц с микроскопическим исследованием для выявления характерных дистрофических изменений.
Причины.
Специалисты полагают, что каждая форма мышечной дистрофии обусловлена отдельным точечным генетическим дефектом, нарушающим способность мышечных клеток синтезировать необходимые белки. Усилия исследователей сосредоточены на поиске дефектов, лежащих в основе заболеваний, и тех отклонений в составе белков, к которым эти дефекты приводят. В настоящее время выявлен ген мышечной дистрофии Дюшенна.
Лечение.
Не существует способов предотвратить или замедлить прогрессирование мышечной слабости при мышечной дистрофии. Терапия направлена главным образом на борьбу с осложнениями, такими, как деформация позвоночника, развивающаяся вследствие слабости мышц спины, или предрасположенность к пневмониям, обусловленная слабостью дыхательных мышц. В этом направлении достигнуты определенные успехи, и качество жизни больных с мышечной дистрофией улучшилось. Сейчас многие больные, несмотря на свой недуг, могут вести полнокровную и продуктивную жизнь.
Проверь себя!
Ответь на вопросы викторины «Здоровье и медицина»
Как звали кардиохирурга, который выполнил первую в мире пересадку сердца от человека человеку?
Препарат для борьбы с мышечной дистрофией Дюшенна улучшает работу энергетических станций клетки
Мышечная дистрофия Дюшенна – наиболее частое нервно-мышечное наследственное заболевание человека. Частота встречаемости составляет 1 на 3500–5000 новорожденных мальчиков в мире. У болезнь ярко выраженный прогрессирующий характер, и уже в подростковом возрасте человек теряет способность к самостоятельному передвижению, а после возникает сердечная и дыхательная недостаточность. С этим заболеванием живут совсем недолго: большинство пациентов умирают в 15–25 лет.
Причина этой тяжелой патологии кроется в мутации гена белка дистрофина. Он отвечает за связь мышечных волокон с клеточным каркасом. Это позволяет поддерживать целостную структуру и функциональность мускулатуры. Вызванный мутациями дефицит этого белка делает мышечные клетки и волокна крайне нестабильными и восприимчивыми к повреждениям. Хотя точные механизмы развития этой патологии все еще требуют дополнительных исследований, уже известно, что большую роль в потере рабочей активности мышечных клеток играет нарушение работы митохондрий. Ранее авторы статьи выяснили, что при болезни Дюшенна в скелетных мышцах появляются серьезные нарушения: митохондрии, производящие энергию и регулирующие транспорт ионов кальция в клетке, начинают работать гораздо менее эффективно. В то же время отмечается, что благодаря митохондриям, клетки сердца, напротив, способны на ранних этапах заболевания сдерживать развитие неблагоприятного сценария. В своей новой работе биологи из республики Марий Эл совместно с коллегами из подмосковного наукограда Пущино изучили влияние терапевтического агента дефлазакорта на работу митохондрий скелетных мышц мышей с точечной мутацией в гене дистрофина, а также здоровых животных, использованных в качестве контроля.
Вызванный мутациями дефицит этого белка делает мышечные клетки и волокна крайне нестабильными и восприимчивыми к повреждениям. Хотя точные механизмы развития этой патологии все еще требуют дополнительных исследований, уже известно, что большую роль в потере рабочей активности мышечных клеток играет нарушение работы митохондрий. Ранее авторы статьи выяснили, что при болезни Дюшенна в скелетных мышцах появляются серьезные нарушения: митохондрии, производящие энергию и регулирующие транспорт ионов кальция в клетке, начинают работать гораздо менее эффективно. В то же время отмечается, что благодаря митохондриям, клетки сердца, напротив, способны на ранних этапах заболевания сдерживать развитие неблагоприятного сценария. В своей новой работе биологи из республики Марий Эл совместно с коллегами из подмосковного наукограда Пущино изучили влияние терапевтического агента дефлазакорта на работу митохондрий скелетных мышц мышей с точечной мутацией в гене дистрофина, а также здоровых животных, использованных в качестве контроля.
«Сегодня одним из путей коррекции мышечной дистрофии Дюшенна является применение глюкокортикоидов, в частности преднизона и его оксазолинового производного дефлазакорта. Ряд других подходов, связанных с подавлением миостатина и применением микродистрофиновой терапии, находятся на стадии испытаний и показывают многообещающие результаты. Однако сейчас дефлазакорт – единственный препарат, официально одобренный Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов в США (FDA) для лечения мышечной дистрофии Дюшенна. Терапия на основе этого глюкокортикоида, обладающего противовоспалительным действием, продлевает двигательную активность пациентов на 2–5 лет, а также улучшает мышечную силу и сердечно-легочную функцию. Кроме того, этот препарат обладает гораздо более «мягкими» побочными эффектами, по сравнению с предшественниками», — рассказывает Михаил Дубинин, руководитель проекта по гранту РНФ, кандидат биологических наук, доцент кафедры биохимии, клеточной биологии и микробиологии Марийского государственного университета (Йошкар-Ола).
В ходе исследования ученые в течение месяца вводили дефлазакорт модельным дистрофин-дефицитным мышам, а также здоровым животным, для того чтобы выявить как положительные, так и отрицательные эффекты такой терапии на работу скелетной мускулатуры и активность митохондрий мышц. Оказалось, что лечение мышей, страдающих мышечной дистрофией, с применением дефлазакорта сопровождается улучшением функциональной активности митохондрий скелетных мышц – они начинают гораздо более активно выполнять свои основные функции, а именно синтезировать АТФ – основную энергетическую молекулу всех живых клеток. Кроме того, они более интенсивно регулируют транспорт ионов кальция, что необходимо для поддерживания сократительной функции скелетной мускулатуры. В конце эксперимента ученые также оценили физическую силу и выносливость мышей с помощью теста на струне – оказалось, что дистрофин-дефицитные мыши, получавшие дефлазакорт, были способны гораздо дольше висеть на струне, держась за нее передними лапами.
«Стоит отметить, что мы обнаружили и побочные эффекты такой терапии. Действительно, митохондрии скелетных мышц мышей, получавших инъекции этого глюкокортикоида, способны гораздо эффективнее поглощать избыток ионов кальция, возникающий в мышечной клетке при дистрофии Дюшенна. Однако в этом случае способность митохондрий удерживать накопленные ионы кальция уменьшалась. Важно отметить, что такой эффект наблюдался и у здоровых животных, получавших дефлазакорт, — говорит Михаил Дубинин. — Поэтому мы полагаем, что со временем такое действие этого глюкокортикоида может приводить к снижению эффективности терапии. Необходима грамотная и своевременная консультация специалистов, а также соблюдение необходимой дозировки при назначении этого препарата».
Исследование проводили сотрудники Марийского государственного университета и Института теоретической и экспериментальной биофизики (ИТЭБ) РАН.
Окулофарингеальная мышечная дистрофия
Окулофарингеальная мышечная дистрофия (ОФМД) — заболевание, которое в мире встречается в двух генетических вариантах: аутосомно-рецессивном и аутосомно-доминантном. Эти варианты болезни являются аллельными и обусловлены различными мутациями в одном гене. Клинические варианты описаны в зависимости от типа наследования. Несмотря на то что, согласно литературным данным, ОФМД относится к более поздним формам мышечных дистрофий (4 – 5 –е десятилетие), имеется клинический пример раннего дебюта ОФМД у пациента в возрасте 23 года, с установленным диагнозом в 31 год.
| Симптомы ОФМД: гипомимия, двусторонний птоз век, наружная офтальмоплегия, «локальная миопатия», поражающая глазодвигательные и поднимающие веки мышцы, а также мышцы-констрикторы глотки (заболевание опасно развитием нарастающей дисфагии), дисфония При длительном течении заболевания — заметная гипотрофия нескольких групп мышц: лица, плечевого пояса, в т.ч. спины, конечностей. Часто появляются бронхо-легочные осложнения. Средний возраст манифестации: 49 ± 1,42 лет |
Особенность окулофарингеальной миодистрофии — наличие в ядрах нитевидных трубочек диаметром 8,5 нм. Аутосомно-доминантный и аутосомно-рецессивный варианты болезни обусловлены мутациями в одном и том же гене — РАВР2 (полиаденилсвязывающем протеине-2; OMIM 602 279), локализованном в области 14q11.2-q13. Основной тип мутаций — короткая экспансия тринуклеотидного повтора GCG в кодирующей части гена. В норме число повторов не превышает 6, однако у 2 % здоровых людей число повторов может достигать 7, что расценивается как проявление нормального полиморфизма. У больных с окулофарингеальной миопатией число повторов увеличено до 8–13.
Электрофореграмма сиквенса GCG – повторов в 1 экзоне гена PABPN1 Результаты электрофореза ДНК на ДНК-анализаторе ABIPRISM 310Тяжесть проявления заболевания зависит от количества повторов. Антиципация, обусловленная увеличением количества повторов, не характерна. Возникновение аутосомно-рецессивного варианта обусловлено гомозиготностью по GCG7-повтору, который является примером аллели-модификатора. Наиболее тяжелый фенотип наблюдался у компаунд-гетерозигот GCG9/GCG7, а также гомозигот по GCG9-повторам. Патогенетически белок РАВР2 является высоконсервативным и содержится в ядре, где участвует в полиаденилировании мРНК. GCG-повторы кодируют включение полиаланинового тракта вблизи N-конца мутантного белка. Считается, что образующиеся в ядре нитевидные структуры представлены удлиненными нитями мутантного белка. Заболевание особенно часто встречается у франко-язычных канадцев и в латиноамериканских семьях на юго-западе США. Оно описано также в большой еврейской семье восточноевропейского происхождения. На сегодняшний день для профилактики ОФМД возможна дородовая диагностика с использованием методов ДНК-анализа.
Антиципация, обусловленная увеличением количества повторов, не характерна. Возникновение аутосомно-рецессивного варианта обусловлено гомозиготностью по GCG7-повтору, который является примером аллели-модификатора. Наиболее тяжелый фенотип наблюдался у компаунд-гетерозигот GCG9/GCG7, а также гомозигот по GCG9-повторам. Патогенетически белок РАВР2 является высоконсервативным и содержится в ядре, где участвует в полиаденилировании мРНК. GCG-повторы кодируют включение полиаланинового тракта вблизи N-конца мутантного белка. Считается, что образующиеся в ядре нитевидные структуры представлены удлиненными нитями мутантного белка. Заболевание особенно часто встречается у франко-язычных канадцев и в латиноамериканских семьях на юго-западе США. Оно описано также в большой еврейской семье восточноевропейского происхождения. На сегодняшний день для профилактики ОФМД возможна дородовая диагностика с использованием методов ДНК-анализа.
ДНК-диагностика ОФМД в 8% ПААГ: (М — маркер PUC19/MspI) 1, 4 — генотип 6/6 GCG-повторов (здоровые). 2, 3 — генотип 6/10 GCG-повторов (больные). |
Опасность окулофарингеальной миодистрофии состоит в прогрессировании с нарастающей дисфагией и требует применения методов паллиативной неврологии. Паллиативная неврология предусматривает зондовое питание или наложение стомы. Эффективного лечения на данный момент нет. Описаны методики рассечения перстнеглоточной мышцы для улучшения глотания, но не предотвращения аспирации. Если птоз мешает зрению, используют специальные скотчевые наклейки на веки, проволочные держатели век, которые крепятся к оправе очков, либо, если нет выраженной слабости мимических мышц, прибегают к хирургическому лечению.
Аутосомно-доминантный вариант заболевания (впервыеописан Victor и соавтор. в 1962 году у 9 членов одной семьи из трех поколений ). Первые симптомы возникают на 4–5-м десятилетиях жизни и в большинстве случаев характеризуются сочетанием дисфагии с прогрессирующим птозом верхних век. По мере прогрессирования заболевания отмечается распространение симптомов мышечной слабости на мышцы плечевого и тазового поясов. Также писана (Satoyoshi & Kinoshita, 1977) семья с аутосомно-доминантной сегрегацией окулофарингеальной миопатии, характеризующейся значительной генерализацией процесса по мере течения болезни. У наблюдаемых больных мышечная слабость распространялась на мышцы лица, шеи, дистальных отделов конечностей, а также анального сфинктера. Описаны единичные больные с наличием пигментной дегенерации сетчатки. Большинство авторов эту форму аутосомно-доминантной миопатии относят к довольно редким, возникающим в зрелом возрасте и медленнотекущим заболеваниям, считают, что клинически болезнь проявляет себя как локальная миопатия. Поражаются мышцы, осуществляющие движения глазных яблок, и мышца, поднимающая верхнее веко. Фарингеальные расстройства обусловлены включением в процесс констрикторов глотки, что затрудняет глотание. Типична симметричность процесса. Начальные признаки болезни появляются в возрасте 30–40 лет: двустороннее опущение верхнего века при ограничении движения глазных яблок. Как правило, на диплопию больные не жалуются. Объяснение этому находят в медленном и симметричном развитии парезов глазодвигательных мышц. Значительно утяжеляют заболевание и ухудшают прогноз фарингеальные симптомы. Начинаясь с дисфагии, они имеют тенденцию к нарушению функций (афагии). Следует иметь в виду существование окулярной миопатии, при которой фарингеальные расстройства не выражены. Этот вариант миопатии рассматривается одними исследователями как самостоятельное заболевание, другими- как дебют окулофарингеальной миопатии.
Первые симптомы возникают на 4–5-м десятилетиях жизни и в большинстве случаев характеризуются сочетанием дисфагии с прогрессирующим птозом верхних век. По мере прогрессирования заболевания отмечается распространение симптомов мышечной слабости на мышцы плечевого и тазового поясов. Также писана (Satoyoshi & Kinoshita, 1977) семья с аутосомно-доминантной сегрегацией окулофарингеальной миопатии, характеризующейся значительной генерализацией процесса по мере течения болезни. У наблюдаемых больных мышечная слабость распространялась на мышцы лица, шеи, дистальных отделов конечностей, а также анального сфинктера. Описаны единичные больные с наличием пигментной дегенерации сетчатки. Большинство авторов эту форму аутосомно-доминантной миопатии относят к довольно редким, возникающим в зрелом возрасте и медленнотекущим заболеваниям, считают, что клинически болезнь проявляет себя как локальная миопатия. Поражаются мышцы, осуществляющие движения глазных яблок, и мышца, поднимающая верхнее веко. Фарингеальные расстройства обусловлены включением в процесс констрикторов глотки, что затрудняет глотание. Типична симметричность процесса. Начальные признаки болезни появляются в возрасте 30–40 лет: двустороннее опущение верхнего века при ограничении движения глазных яблок. Как правило, на диплопию больные не жалуются. Объяснение этому находят в медленном и симметричном развитии парезов глазодвигательных мышц. Значительно утяжеляют заболевание и ухудшают прогноз фарингеальные симптомы. Начинаясь с дисфагии, они имеют тенденцию к нарушению функций (афагии). Следует иметь в виду существование окулярной миопатии, при которой фарингеальные расстройства не выражены. Этот вариант миопатии рассматривается одними исследователями как самостоятельное заболевание, другими- как дебют окулофарингеальной миопатии.
Аутосомно-рецессивный вариант заболевания (впервые описан Fried и соавт. в 1975 году у двух сестер, родившихся от кровнородственного брака). Для этой формы болезни характерно более раннее начало и вовлечение в процесс дистальных групп мышц конечностей. При (ЭМГ) электромиографическом обследовании больных с ОФМД определяют первично-мышечный характер поражения. При морфологическом исследовании выявляют нитевидные образования в ядрах скелетных мышц. Эти нити имеют ветвящуюся трубчатую структуру и иногда поперечно исчерчены. Наряду с этим отмечаются атрофические изменения в мышечных волокнах 1-го типа. При электронном микроскопировании обнаруживается увеличение размеров митохондрий с наличием в них крестовидных включений. Также могут быть обнаружены вакуоли, при электронной микроскопии в них видны обрывки мембран, скопления гликогена и другие неспецифичные остатки лизосомного происхождения.
При (ЭМГ) электромиографическом обследовании больных с ОФМД определяют первично-мышечный характер поражения. При морфологическом исследовании выявляют нитевидные образования в ядрах скелетных мышц. Эти нити имеют ветвящуюся трубчатую структуру и иногда поперечно исчерчены. Наряду с этим отмечаются атрофические изменения в мышечных волокнах 1-го типа. При электронном микроскопировании обнаруживается увеличение размеров митохондрий с наличием в них крестовидных включений. Также могут быть обнаружены вакуоли, при электронной микроскопии в них видны обрывки мембран, скопления гликогена и другие неспецифичные остатки лизосомного происхождения.
Распространенность ОФМД у якутов в 10 раз выше распространенности описанной в мире. ОФМД получила накопление на территории улусов центральной Якутии и вилюйских улусов, особенно в Усть-Алданском улусе и городе Якутске.Выявлены мажорные мутации, характерные для якутской популяции: экспансия (GCG)10 в гене PÁBPN1 для окулофарингеальной миодистрофии. Распространенность аутосомно-доминантной окулофарингеальной миодистрофии в PC (Я) среди якутов составляет 11,1 на 100 тыс. населения с преимущественным накоплением в центральной и вилюйской группах улусов Якутии. Единственная молекулярно-генетическая причина всех случаев ОФМД в якутской популяции — мутация (GCG)m в 1 экзоне гена PABPN1. Выявленный единственный гаплотип является гаплотипом основателя.
Литература по ОФМД в якутской популяции:
- Внедрение ДНК-диагностики окулофарингеальной миодистрофии в практику медико-генетического консультирования Республики Саха (Якутия) // Куртанов Х.А., Максимова Н.Р., Стапанова С.К. и др. // Якутский медицинский журнал. 2008. №4. С. 43-46.
- Генетико-эпидемиологические и социально-экономические аспекты наследственной этноспецифической патологии в Якутии // Максимова Н.Р., Сухомясова A.JI., Гуринова Е.Е. и др. // Медицинская генетика. 2008. Т. 7. №10. С. 35-43.
- ДНК-диагностика моногенных заболеваний в Республике Саха (Якутия)//Максимова Н. Р., Сухомясова A.JL, Томский М.И. // Илин. 2009. №3. С. 54-56.
- Клинико-генеалогическая и молекулярно-генетическая характеристика окулофарингеальной миодистрофии в Республике Саха (Якутия)//Максимова Н.Р., Николаева И.А., Коротов М.Н. и др. // Журнал неврологии и психиатрии им. С.С. Корсакова. 2008. Т. 108. №6. С. 52-60.
- Клинико-генеалогическая и молекулярно-генетическая характеристика этноспецифических форм наследственной патологии у якутов // Максимова Надежда Романовна //автореф. дисс. на соиск. уч. степ. доктора медицинских наук // Томск – 2009
- Клинико-генеалогическое и молекулярно-генетическое исследование окулофарингеальной миодистрофии в Республике Саха (Якутия) // Максимова Н.Р., Николаева И.А., Коротов М.Н. и др. / C6.VIII научно-практической конференции «Генетика человека и патология». Томск: Изд-во «Печатная мануфактура», 2007. С. 160-161.
- Молекулярно-генетические методы диагностики наследственных моногенных болезней в РБ№1-НЦМ PC (Я) //Степанова С.К., Кононова С.К., Федорова С.А., Максимова Н.Р. и др. // Тез. межрегиональной научно-практической конференции «Молекулярно-клеточные аспекты патологии человека на Севере». Якутск: ЯНЦ СО РАМН, 2007. С. 49-51.
- Наследственные болезни нервной системы в Республике Саха (Якутия) // Николаева И.А., Коротов М.Н., Гуринова Е.Е., Степанова С.К., Максимова Н.Р. и др. // Якутский медицинский журнал. 2009. №2. С. 52-54.
- Наследственные болезни у якутов // В.П.Пузырев, Н.Р.Максимова
- Окулофарингеальная миодистрофия в Республике Саха (Я) // Максимова Н.Р., Коротов М.Н., Николаева И.А. и др. // Тез. III международной научно-практической конференции «Проблемы вилюйского энцефаломиелита и других нейродегенеративных заболеваний в Якутии». Якутск: «Копиртех-сервис», 2006. С. 22-23.
- миодистрофия в Республике Саха (Якутия): клинические и молекулярно-генетические аспекты // Максимова Н.Р., Николаева И.А., Коротов М.Н. и др. // Тез. П межрегиональной научно-практической конференции «Экология и здоровье человека на Севере». Якутск: ЯНЦ СО РАМН, 2007. С. 34.
- Полиморфизм локуса ОФМД в популяциях Якутии // Куртанов Х.А., Максимова Н.Р., Марусин A.B., Степанов В.А. // Якутский медицинский журнал. 2009. №2. С. 54-58.
- Этноспецифические наследственные болезни у якутов // Максимова Н.Р., Пузырев В.П / Тез. межрегиональной научно-практической конференции «Здоровье детей на Севере». Якутск: ЯНЦ СО РАМН, 2008. С. 91-94.
- Этноспецифическая наследственная патология в РС (Я)// Максимова Н.Р., Сухомясова А.Л., Ноговицына А.Н., Пузырев В.П.
- Литература по ОФМД в других популяциях: ДНК-диагностика случая окулофарингеальной миодистрофии в России // Чухрова А.Л., Федотов В.П., Поляков А.В. // Медицинская генетика, 2003 г., Т.2. №10 «Тезисы Всероссийской научно-практической конф. «Соврем. достиж. клинич. генетики», Москва, 25-27 ноября 2003 г., стр. 446
- Клинико-молекулярно-генетическая классификация мышечных дистрофий (научный обзор с комментариями // Казаков В.М. // Неврол. журнал. — 2001. — № 3. — С. 47-52.
- Молекулярная неврология. Часть 1. Заболевания нервно-мышечной системы. // Горбунова В.Н., Савельева-Васильева Е.А., Красильников В.В. // СПб.: Интермедика, 2000. — 320 с.
- Ранняя клинико-инструментальная диагностика и терапия быстро- и медленнопрогрессирующих мышечных дистрофий и амиотрофий // 3. Евтушенко С.К., Шаймурзин М.Р., Евтушенко О.С., Евтушенко Л.Ф., Дегонская Е.В., Евтушенко И.С., Сохань Д.А.. // Международный неврологический журнал. — 2007. — № 4(14). — С. 47.
- Справочник по формулированию клинического диагноза болезней нервной системы / Под ред. В.Н. Штока, О.С. Левина. — М.: ООО «Медицинское информационное агентство», 2006. — 520 с.
- Brais B., Bouchard J.-P., Xie Y.-G., Rochefort D.L., Chretien N., Tome F.M.S., Lafreniere R.G., Rommens J.M., Uyama E., Nohira O., Blumen S., Korcyn A.D., Heutink P., Mathieu J., Duran-ceau A., Codere F., Fardeau M., Rouleau G.A. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy // Nature Genet. — 1998. — 18. — 164-167.
- Brais B., Xie Y.-G., Sanson M., Morgan K., Weissenbach J., Korczyn A.D., Blumen S.C., Fardeau M., Tome F.M.S., Bou-chard J.-P., Rouleau G.A. The oculopharyngeal muscular dystrophy locus maps to the region of the cardiac alpha and beta myosin heavy chain genes on chromosome 14q11.2-q13 // Hum. Molec. Genet. — 1995. — 4. — 429-434.
- Fried K., Arlozorov A., Spria R. Autosomal recessive oculopharyngeal muscular dystrophy // J. Med. Genet. — 1975. — 12. — 416-418.
- Goh K.J., Wong K.T., Nishino I., Minami N., Nonaka I. Oculopharyngeal muscular dystrophy with PABPN1 mutation in a Chinese Malaysian woman // Neuromusc. Disord. 2005. — 15. — 262-264.
- Hino H., Araki K., Uyama E., Takeya M., Araki M., Yoshinobu K., Miike K., Kawazoe Y., Maeda Y., Uchino M., Yamamura K. Myopathy phenotype in transgenic mice expressing mutated PABPN1 as a model of oculopharyngeal muscular dystrophy // Hum. Molec. Genet. — 2004. — 13. — 181-190.
- Neuromuscular disorders: gene location // Neuromusc. Disord. — 1999. — Vol. 9, № 5. — P. 1-8.
- Robinson D.O., Wills A.J., Hammans S.R., Read S.P., Sillibourne J. Oculopharyngeal muscular dystrophy: a point mutation which mimics the effect of the PABPN1 gene triplet repeat expansion mutation // J. Med. Genet. — 2006. — 43. — e23 (Note: Electronic Article).
- Victor M., Hayes R., Adams R.D. Oculopharyngeal muscular dystrophy. A familial disease of late life characterized by dysphagia and progressive ptosis of the eyelids // New Eng. J. Med. — 1962. — 267. — 1267-1272.
 Р., Сухомясова A.JL, Томский М.И. // Илин. 2009. №3. С. 54-56.
Р., Сухомясова A.JL, Томский М.И. // Илин. 2009. №3. С. 54-56. Якутск: ЯНЦ СО РАМН, 2007. С. 34.
Якутск: ЯНЦ СО РАМН, 2007. С. 34. — 1998. — 18. — 164-167.
— 1998. — 18. — 164-167.Мышечная дистрофия: симптомы, диагностика и лечение
Что такое мышечная дистрофия?
Мышечная дистрофия — это группа заболеваний, при которых мышцы со временем становятся слабее и менее гибкими. Это вызвано проблемой в генах, которые контролируют, как тело поддерживает здоровье мышц. У некоторых людей болезнь начинается в раннем детстве. У других нет никаких симптомов, пока они не станут подростками или взрослыми людьми среднего возраста.
Каким образом мышечная дистрофия влияет на вас или вашего ребенка, зависит от ее вида.Состояние большинства людей со временем ухудшается, и некоторые люди могут потерять способность ходить, говорить или заботиться о себе. Но это случается не со всеми. Другие люди могут жить долгие годы с легкими симптомами.
Существует более 30 видов мышечной дистрофии, и каждый из них отличается в зависимости от:
- Гены, вызывающие ее
- Мышцы, поражающие ее
- Возраст, когда симптомы впервые появляются
- Как быстро болезнь ухудшается
Люди обычно болеют одной из девяти основных форм болезни:
- Мышечная дистрофия Дюшенна (МДД) является наиболее распространенной формой.Это в основном поражает мальчиков и начинается в возрасте от 3 до 5 лет.
- Мышечная дистрофия Беккера похожа на мышечную дистрофию Дюшена, за исключением более легкой степени. Он также поражает мальчиков, но симптомы проявляются позже — в возрасте от 11 до 25 лет.
- Миотоническая мышечная дистрофия — наиболее распространенная форма у взрослых. Люди, у которых он есть, не могут расслабить мышцы после сокращения. Это может повлиять как на мужчин, так и на женщин, и обычно начинается, когда людям за двадцать.
- Врожденная мышечная дистрофия начинается при рождении или вскоре после него.
- Мышечная дистрофия конечностей-пояса часто начинается в подростковом или 20-летнем возрасте.
- Facioscapulohumeral мышечная дистрофия поражает мышцы лица, плеч и предплечий. Это может повлиять на кого угодно, от подростков до взрослых в возрасте от 40 лет.
- Дистальная мышечная дистрофия поражает мышцы рук, ног, кистей и стоп. Обычно она возникает в более зрелом возрасте, в возрасте от 40 до 60 лет.
- Глоточная мышечная дистрофия начинается у человека в возрасте 40–50 лет.Это вызывает слабость мышц лица, шеи и плеч и опущение век (птоз) с последующим затруднением глотания (дисфагия).
- Мышечная дистрофия Эмери-Дрейфуса поражает в основном мальчиков, обычно начиная с 10 лет. Люди с этой формой часто имеют проблемы с сердцем наряду с мышечной слабостью.

Существует множество методов лечения, которые могут помочь сохранить мышцы сильными и гибкими, и ученые тоже ищут новые. Важно получить необходимое лечение и найти поддержку.
Причины
Мышечная дистрофия может передаваться в семье, или вы можете быть первым в своей семье, у кого она есть. Это состояние вызвано проблемами в ваших генах.
Гены содержат информацию, необходимую вашим клеткам для выработки белков, контролирующих все функции организма. Когда у гена есть проблема, ваши клетки могут вырабатывать неправильный белок, неправильное его количество или поврежденный белок.
Вы можете получить мышечную дистрофию, даже если ни один из ваших родителей не болел.Это происходит, когда один из ваших генов дефектен сам по себе. Но так бывает редко.
У людей с мышечной дистрофией поврежденные гены — это те, которые вырабатывают белки, которые делают мышцы здоровыми и сильными. Например, люди с мышечной дистрофией Дюшенна или Беккера вырабатывают слишком мало белка, называемого дистрофином, который укрепляет мышцы и защищает их от травм.
Симптомы
Для большинства типов мышечной дистрофии симптомы начинают проявляться в детстве или в подростковом возрасте.Как правило, дети с этим заболеванием:
- Часто падают
- Имеют слабые мышцы
- Мышечные судороги
- Проблемы с подъемом, подъемом по лестнице, бегом или прыжками
- Ходите на носках или переваливайте ногами
Некоторые также будут иметь следующие симптомы:
- Искривленный позвоночник (так называемый сколиоз)
- Опущенные веки
- Проблемы с сердцем
- Проблемы с дыханием или глотанием
- Проблемы со зрением
- Слабость лицевых мышц
Получение диагноза
Вашему врачу необходимо будет проверить различные части тела вашего ребенка, чтобы узнать, есть ли у них мышечная дистрофия. Они начнут с общего медицинского осмотра. Они также зададут вам вопросы об истории болезни вашей семьи и о симптомах, которые вы замечаете у своего ребенка. Они могут спросить:
Они начнут с общего медицинского осмотра. Они также зададут вам вопросы об истории болезни вашей семьи и о симптомах, которые вы замечаете у своего ребенка. Они могут спросить:
- Какие мышцы доставляют им проблемы?
- Им трудно ходить или заниматься своими обычными делами?
- Как давно это происходит?
- Есть ли у кого-нибудь в вашей семье мышечная дистрофия? Какой?
Они также могут задать вам вопросы о том, как ваш ребенок играет, двигается и говорит, а также как он ведет себя дома и в школе.
Продолжение
Врач может использовать различные тесты для проверки условий, которые могут вызвать мышечную слабость.
- Анализы крови. Они проверяют уровень определенных ферментов, которые выделяют мышцы при их повреждении.
- Электромиография, или ЭМГ . Ваш врач наложит маленькие иглы, называемые электродами, на разные части тела вашего ребенка и попросит его медленно согнуть и расслабить мышцы. Электроды прикреплены проводами к устройству, измеряющему электрическую активность.
- Биопсия мышц. С помощью иглы врач удаляет небольшой кусочек мышечной ткани ребенка. Они будут смотреть на это под микроскопом, чтобы определить, какие белки отсутствуют или повреждены. Этот тест может показать, какой тип мышечной дистрофии может быть у вашего ребенка.
- Тесты мышечной силы, рефлексов и координации. Помогают врачам исключить другие проблемы с нервной системой.
- Электрокардиограмма или ЭКГ. Он измеряет электрические сигналы, исходящие от сердца, и сообщает, насколько быстро бьется сердце вашего ребенка и его ритм здоровый.
- Визуализация может показать качество и количество мышц в теле вашего ребенка. Они могут получить:
- МРТ или магнитно-резонансная томография. Он использует мощные магниты и радиоволны, чтобы делать снимки своих органов.
- Ультразвук, , который использует звуковые волны для создания изображений внутренней части тела.

Врачи также могут проанализировать образец своей крови, чтобы найти гены, вызывающие мышечную дистрофию. Генетические тесты могут помочь диагностировать заболевание, но они также важны для людей с семейным анамнезом заболевания, которые планируют создать семью.Вы можете поговорить со своим врачом или консультантом по генетике, чтобы узнать, что результаты этого теста значат для вас и ваших детей.
Вопросы для врача
Вам нужно узнать как можно больше о состоянии вашего ребенка, чтобы узнать, как он может оставаться максимально здоровым. Вы можете спросить:
- Какая у них мышечная дистрофия?
- Нужны ли им еще тесты?
- Нужно ли нам обращаться к другим врачам?
- Как болезнь повлияет на их жизнь?
- Какие виды лечения доступны?
- Как они будут себя чувствовать?
- Что я могу сделать, чтобы их мышцы оставались сильными?
- Существуют ли какие-либо клинические испытания, которые были бы им полезны?
- Заболеют ли другие мои дети мышечной дистрофией?
Лечение
На данный момент лекарства от болезни нет.Но существует множество методов лечения, которые могут улучшить симптомы и облегчить жизнь вам и вашему ребенку.
Ваш врач порекомендует лечение в зависимости от типа мышечной дистрофии вашего ребенка. Вот некоторые из них:
- Физическая терапия использует различные упражнения и растяжки, чтобы мышцы оставались сильными и гибкими.
- Трудотерапия учит вашего ребенка, как максимально использовать то, на что способны его мышцы. Терапевты также могут показать им, как пользоваться инвалидными колясками, скобами и другими приспособлениями, которые могут помочь им в повседневной жизни.
- Логопед научит их более простым способам говорить, если у них слабые мышцы горла или лица.
- Респираторная терапия может помочь, если у вашего ребенка проблемы с дыханием. Они узнают, как облегчить дыхание, или попросят в помощь машины.
- Лекарства могут облегчить симптомы. Они включают:
- Этеплирсен (Exondys 51), голодирсен (Vyondys53) и витоларсен (Вилтепсо) для лечения МДД. Это инъекционные препараты, которые помогают лечить людей с определенной мутацией гена, приводящей к МДД, в частности, за счет увеличения выработки дистрофина.Поговорите с врачом вашего ребенка о возможных побочных эффектах.
- Противосудорожные препараты , уменьшающие мышечные спазмы.
- Лекарства от кровяного давления , которые помогают при сердечных заболеваниях.
- Лекарства, подавляющие иммунную систему организма , , называемые иммунодепрессантами; они могут замедлить повреждение мышечных клеток.
- Стероиды , такие как преднизон и дефказакорт (Эмфлаза), замедляют повреждение мышц и могут помочь вашему ребенку лучше дышать.Они могут вызывать серьезные побочные эффекты, такие как слабость костей и повышенный риск инфекций.
- Креатин, — химическое вещество, обычно содержащееся в организме, которое помогает снабжать мышцы энергией и улучшает силу у некоторых людей. Спросите врача, подходят ли ему эти добавки.
- Хирургия может помочь с различными осложнениями мышечной дистрофии, такими как проблемы с сердцем или проблемы с глотанием.
 Они узнают, как облегчить дыхание, или попросят в помощь машины.
Они узнают, как облегчить дыхание, или попросят в помощь машины.Ученые также ищут новые способы лечения мышечной дистрофии в клинических испытаниях.Эти испытания проверяют новые лекарства, чтобы убедиться, что они безопасны и работают. Часто они позволяют людям попробовать новое лекарство, доступное далеко не каждому. Ваш врач может сказать вам, подходит ли одно из этих испытаний для вашего ребенка.
Забота о ребенке
Трудно, когда ваш ребенок теряет силы и не может делать то, что могут делать другие дети. Мышечная дистрофия — это проблема, но она не должна мешать вашему ребенку получать удовольствие от жизни.
Есть много вещей, которые вы можете сделать, чтобы помочь им почувствовать себя сильнее и получить от жизни максимум удовольствия.
- Правильно питайтесь. Здоровая, хорошо сбалансированная диета полезна для вашего ребенка в целом. Это также важно для поддержания здорового веса, который может облегчить проблемы с дыханием и другие симптомы. Если им трудно жевать или глотать, посоветуйтесь с диетологом о продуктах, которые, возможно, будет легче съесть.
- Оставайтесь активными. Физические упражнения могут улучшить мышечную силу вашего ребенка и улучшить его самочувствие. Попробуйте занятия с низкой нагрузкой, например плавание.
- Высыпайтесь. Спросите своего врача или терапевта о некоторых кроватях или подушках, которые могут сделать вашему ребенку более комфортный и отдохнувший.
- Используйте подходящие инструменты. Инвалидные коляски, костыли или электросамокаты могут помочь вашему ребенку, если у него проблемы с ходьбой.
Болезнь, скорее всего, сильно повлияет на вашу семью. Помните, что это нормально — обращаться за помощью к врачу, психологу, семье или друзьям с любым стрессом, грустью или гневом, которые вы можете испытывать. Группы поддержки также являются хорошим местом для общения с другими людьми, которые жили с мышечной дистрофией.Они могут помочь вашему ребенку общаться с такими же людьми, как они, и дать вам и вашей семье совет и понимание.
Чего ожидать
Мышечная дистрофия у всех разная. Некоторые дети могут очень медленно терять мышечную силу, что дает им и их семьям время, чтобы приспособиться к изменениям. Другим будет хуже быстрее. Многим людям с этим заболеванием в какой-то момент понадобятся инвалидные коляски и помощь в повседневной жизни, но это не всегда так.
Поговорите со своим врачом о мышечной дистрофии вашего ребенка.Вместе вы сможете составить для них наилучший план лечения и получить необходимую поддержку для своей семьи.
Получение поддержки
Чтобы узнать больше о мышечной дистрофии или найти группу поддержки в вашем районе, посетите веб-сайт Ассоциации мышечной дистрофии.
Мышечная дистрофия — NHS
Мышечные дистрофии (МД) — это группа наследственных генетических состояний, которые постепенно вызывают ослабление мышц, что приводит к возрастающему уровню инвалидности.
MD — это прогрессирующее состояние, что означает, что со временем оно ухудшается. Он часто начинается с воздействия на определенную группу мышц, а затем затрагивает более широкие мышцы.
Некоторые типы MD в конечном итоге влияют на сердце или мышцы, используемые для дыхания, после чего состояние становится опасным для жизни.
Нет лекарства от MD, но лечение может помочь справиться со многими симптомами.
Что вызывает мышечную дистрофию?
MD вызван изменениями (мутациями) генов, отвечающих за структуру и функционирование мышц человека.
Мутации вызывают изменения в мышечных волокнах, которые мешают мышцам функционировать. Со временем это приводит к увеличению инвалидности.
Мутации часто передаются по наследству от родителей. Если в вашем семейном анамнезе есть MD, ваш терапевт может направить вас на генетическое тестирование и консультацию, чтобы оценить ваш риск развития этого заболевания или рождения ребенка с MD, а также обсудить доступные вам варианты.
Узнайте больше о причинах MD и генетическом тестировании на MD.
Виды мышечной дистрофии
Существует много разных типов MD, каждый из которых имеет несколько разные симптомы. Не все типы вызывают тяжелую инвалидность, и многие не влияют на продолжительность жизни.
Некоторые из наиболее распространенных типов MD включают:
- MD Duchenne MD — одна из самых частых и тяжелых форм, обычно поражает мальчиков в раннем детстве; люди с этим заболеванием обычно доживают до 20-30 лет
- миотоническая дистрофия — разновидность МД, которая может развиться в любом возрасте; На продолжительность жизни не всегда влияет, но люди с тяжелой формой миотонической дистрофии могли сократить жизнь
- facioscapulohumeral MD — тип MD, который может развиться в детстве или в зрелом возрасте; прогрессирует медленно и обычно не опасно для жизни
- Becker MD — близкий родственник Duchenne MD, но развивается позже в детстве и протекает в меньшей степени; на продолжительность жизни обычно не так сильно влияет
- конечность-пояс MD — группа состояний, которые обычно развиваются в позднем детстве или в раннем взрослом возрасте; некоторые варианты могут быстро прогрессировать и быть опасными для жизни, тогда как другие развиваются медленно
- окулофарингеальный MD — тип MD, который обычно не развивается, пока человеку не исполнится от 50 до 60 лет, и не влияет на ожидаемую продолжительность жизни
- Emery-Dreifuss MD — тип MD, развивающийся в детстве или в раннем взрослом возрасте; большинство людей с этим заболеванием доживут как минимум до среднего возраста
Подробнее о видах МД и диагностировании МД.
Кто страдает мышечной дистрофией?
В Великобритании около 70 000 человек имеют MD или родственное заболевание.
Дюшенн MD является наиболее распространенным типом MD. В Великобритании ежегодно рождается около 100 мальчиков с диагнозом Дюшенна, а в Великобритании одновременно проживает около 2500 человек с этим заболеванием.
Миотонический MD является вторым по распространенности типом MD, которым страдает примерно 1 человек из 8000.
Считается, чтоFacioscapulohumeral MD поражает примерно 1 человека из 20 000 в Великобритании, что делает его третьим по распространенности MD.
Диагностика мышечной дистрофии
Для диагностики различных типов МД можно использовать множество различных методов. Возраст, в котором диагностируется состояние, будет варьироваться в зависимости от того, когда впервые начинают проявляться симптомы.
Диагностика включает некоторые или все следующие этапы:
- исследование любых симптомов
- , обсуждая любую семейную историю MD
- медицинский осмотр
- Анализы крови
- Электрические пробы нервов и мышц
- Биопсия мышцы (при которой для исследования отбирается небольшой образец ткани)
Обратитесь к терапевту, если у вас или у вашего ребенка есть какие-либо симптомы MD.При необходимости ваш терапевт может направить вас в больницу для проведения дополнительных анализов.
Лечение мышечной дистрофии
Нет лекарства от MD, но ряд методов лечения может помочь с физическими недостатками и проблемами, которые могут развиться. Сюда могут входить:
- Помощь при передвижении, включая упражнения, физиотерапию и физические вспомогательные средства Группы поддержки
- — чтобы справиться с практическим и эмоциональным воздействием MD
- хирургия — для исправления постуральных деформаций, например сколиоза
- лекарства, такие как стероиды для улучшения мышечной силы или ингибиторы АПФ и бета-блокаторы для лечения сердечных заболеваний
Новое исследование изучает способы исправления генетических мутаций и поврежденных мышц, связанных с MD. В настоящее время существуют многообещающие клинические испытания MD Duchenne.
В настоящее время существуют многообещающие клинические испытания MD Duchenne.
Подробнее о лечении MD.
Информация о вас
Если у вас есть MD, ваша клиническая бригада передаст информацию о вас в Национальную службу регистрации врожденных аномалий и редких заболеваний (NCARDRS).
Это помогает ученым искать более эффективные способы профилактики и лечения этого состояния. Вы можете отказаться от регистрации в любое время.
Узнайте больше о реестре.
Группы поддержки
MD может повлиять на вас как эмоционально, так и физически. Группы поддержки и организации могут помочь вам понять и смириться с вашим заболеванием. Они также могут предоставить полезные советы и поддержку тем, кто заботится о людях с МД.
Есть несколько национальных благотворительных организаций, которые предлагают поддержку людям, страдающим MD, например, Muscle Help Foundation и Muscular Dystrophy UK. Вы также можете спросить у своего терапевта или других лечащих вас медицинских работников о местных группах поддержки.
Есть также несколько национальных групп поддержки, которые продвигают исследования или оказывают поддержку конкретным типам MD, такие как Action Duchenne, Duchenne UK и Группа поддержки миотонической дистрофии.
Последняя проверка страницы: 24 мая 2018 г.
Срок следующей проверки: 24 мая 2021 г.
Мышечная дистрофия | Сидарс-Синай
Не то, что вы ищете?Обзор
Мышечная дистрофия — это группа генетических заболеваний, вызывающих прогрессирующую слабость мышц тела.Некоторые типы мышечной дистрофии проявляются симптомами в раннем детстве, а другие — в зрелом возрасте. Различные группы мышц также могут быть затронуты в зависимости от типа мышечной дистрофии.
Мышечная дистрофия Дюшенна — наиболее распространенная форма мышечной дистрофии у детей.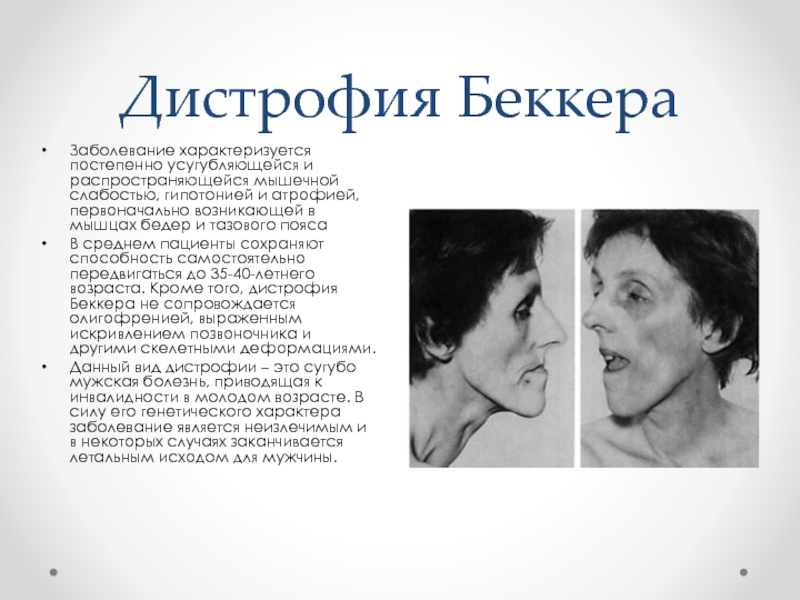 Обычно он появляется у мальчиков 3–6 лет и быстро ухудшается.
Обычно он появляется у мальчиков 3–6 лет и быстро ухудшается.
Мышечная дистрофия Беккера имеет симптомы, аналогичные мышечной дистрофии Дюшенна. Однако симптомы чаще всего начинаются в подростковом возрасте до 25 лет и медленно прогрессируют.
Симптомы
Различные типы мышечной дистрофии начинают проявлять симптомы в разном возрасте, и, в зависимости от типа мышечной дистрофии, поражаются разные группы мышц по всему телу.
Симптомы могут включать:
- Прогрессирующая мышечная слабость и истощение (атрофия)
- Перевалка
- Сложный подъем по лестнице
- Затруднение при вставании из положения лежа или сидя
- Повторное падение
- Искривление позвоночника
- Истощение мышц бедра
- Аномальное увеличение икр
- Проблемы с дыханием или глотанием
- Конечности втягиваются внутрь и фиксируются в положении
- Увеличение сердца
Помимо мышечных дистрофий Дюшенна и Беккера, существуют и другие, менее распространенные типы:
- Фациоскапуло-плечевая мышечная дистрофия (синдром Ландузи-Дежерина), поражающая в основном мышцы лица, плеч и рук
- Конечностно-поясная мышечная дистрофия, сначала поражающая бедра и плечи
- Миотоническая мышечная дистрофия (MMD или болезнь Штейнерта), которая вызывает неспособность расслабить мышцы и обычно сначала поражает лицевые мышцы
- Окулофарингеальная мышечная дистрофия, обычно возникающая в среднем возрасте со слабостью мышц глаз, лица и горла, вызывающая опущение век и проблемы с глотанием
Причины и факторы риска
Дефектные гены являются причиной мышечной дистрофии.Специфическое генное заболевание было обнаружено для наиболее распространенных мышечных дистрофий. Например, дефектный ген дистрофина, белка, который помогает сохранить неповрежденными мышечные клетки, является причиной мышечной дистрофии Дюшенна.
Для менее распространенных форм мышечной дистрофии исследователи все еще пытаются найти конкретный дефект гена, который вызывает заболевание. Хотя большинство форм мышечной дистрофии передаются по наследству, некоторые из них возникают в результате спонтанной генной мутации без какой-либо семейной истории болезни.
Диагностика
Чтобы диагностировать любую форму мышечной дистрофии, врач тщательно изучит историю болезни и проведет медицинский осмотр. Также можно заказать некоторые диагностические тесты.
Анализ крови покажет, вытекает ли фермент креатинкиназа из мышечных клеток, вызывая аномально высокий уровень в крови. Хотя высокий уровень креатинкиназы в крови не обязательно подтверждает наличие у пациента мышечной дистрофии, это признак мышечной болезни.
Электромиография и исследования нервной проводимости позволяют измерять электрическую активность мышц и нервной системы для выявления наличия, локализации и степени заболеваний, которые могут повредить мышечную ткань.
Биопсия мышцы, при которой небольшой кусочек мышцы удаляется для исследования под микроскопом, обычно проводится для подтверждения состояния. Под микроскопом мышца после положительной биопсии обычно показывает мертвую ткань и аномально большие мышечные волокна. На поздних стадиях мышечной дистрофии жир и другие ткани заменяют мертвую мышечную ткань.
Генетическое тестирование может быть выполнено для определения генных мутаций, вызвавших мышечную дистрофию.
Лечение
В настоящее время не существует лекарств от мышечной дистрофии. Лечебная физкультура и упражнения помогают предотвратить постоянное сокращение мышц вокруг суставов и избежать искривления позвоночника. Иногда требуется операция, чтобы освободить напряженные болезненные мышцы. Дыхательные упражнения помогают отсрочить ослабление дыхательных мышц.
Некоторые симптомы можно вылечить, а прогрессирование можно замедлить с помощью лекарств. Преднизон, мощный кортикостероидный препарат, в настоящее время используется для временного облегчения мышечной слабости и замедления их повреждения, а также для улучшения дыхательной функции. При некоторых типах мышечной дистрофии могут возникнуть проблемы с сердцем, которые можно лечить с помощью лекарств или кардиостимулятора.
Преднизон, мощный кортикостероидный препарат, в настоящее время используется для временного облегчения мышечной слабости и замедления их повреждения, а также для улучшения дыхательной функции. При некоторых типах мышечной дистрофии могут возникнуть проблемы с сердцем, которые можно лечить с помощью лекарств или кардиостимулятора.
Исследователи изучают генную терапию, которая позволила бы мышцам вырабатывать дистрофин (от мышечных дистрофий Дюшенна и Беккера), а также другие методы лечения, чтобы найти лекарство от всех мышечных дистрофий.
Не то, что вы ищете?Мышечная дистрофия (для родителей) — Nemours KidsHealth
[Перейти к содержанию] Открыть поиск- для Родители
- Родительский сайт
- Sitio para padres
- Здравоохранение
- Рост и развитие
- Инфекции
- Заболевания и состояния
- Беременность и младенец
- Питание и фитнес
- Эмоции и поведение
- Школьная и семейная жизнь
- Первая помощь и безопасность
- Врачи и больницы
- Видео
- Рецепты
- Закрыть для родителей навиг.
- для детей
- Детский сайт
- Sitio para niños
- Как работает тело
- Половое созревание и взросление
- Сохранение здоровья
- Остаться в безопасности
- Рецепты и кулинария
- Проблемы со здоровьем
- Болезни и травмы
- Расслабься и расслабься
- Люди, места и вещи, которые помогают
- Чувства
- Ответы экспертов Q&A
- Фильмы и другое
- Закрыть детское навигатор
- для подростков
- Подростковая площадка
- Sitio para adolescentes
- Кузов
- Разум
- Сексуальное здоровье
- Еда и фитнес
- Заболевания и состояния
- Инфекции
- Наркотики и алкоголь
- Школа и работа
- Спорт
- Ответы экспертов (вопросы и ответы)
- Безопасность
Мышечные дистрофии у детей | Детская национальная больница
Что такое мышечная дистрофия?
Мышечная дистрофия (МД) — это широкий термин, который описывает генетическое (наследственное) заболевание мышц. Мышечная дистрофия приводит к тому, что мышцы тела становятся очень слабыми. Мышцы разрушаются и со временем заменяются жировыми отложениями.
Мышечная дистрофия приводит к тому, что мышцы тела становятся очень слабыми. Мышцы разрушаются и со временем заменяются жировыми отложениями.
К другим проблемам со здоровьем, обычно связанным с мышечной дистрофией, относятся следующие:
- Проблемы с сердцем
- Сколиоз. Боковое или боковое искривление и вращение костей спины (позвонков), создающее впечатление, что человек наклоняется в сторону.
- Ожирение
Наиболее распространенными формами мышечной дистрофии являются мышечная дистрофия Дюшенна (МДД) и мышечная дистрофия Беккера.Эти две формы очень похожи, но мышечная дистрофия Беккера менее серьезна, чем МДД. Девочки редко страдают любой из этих двух форм мышечной дистрофии.
Что вызывает мышечную дистрофию?
Мышечная дистрофия Дюшенна — это генетическое заболевание, что означает, что оно передается по наследству. Наши гены определяют наши черты характера, такие как цвет глаз и группа крови. Гены содержатся в клетках нашего тела на палочкообразных структурах, называемых хромосомами. Обычно в каждой клетке нашего тела 46 хромосом, или 23 пары.Первые 22 пары являются общими для мужчин и женщин, в то время как последняя пара определяет пол и называется парой половых хромосом: у женщин есть две X-хромосомы, а у мужчин — одна X и одна Y-хромосома.
Мышечная дистрофия Дюшенна вызывается Х-сцепленным рецессивным геном. «Х-сцепленный» означает, что ген, вызывающий признак или нарушение, расположен на Х-хромосоме. Гены на X-хромосоме могут быть рецессивными или доминантными, и их экспрессия у женщин и мужчин не одинакова, потому что гены на Y-хромосоме не точно спариваются с генами на X.Х-сцепленные рецессивные гены экспрессируются у женщин только при наличии двух копий гена (по одной на каждой Х-хромосоме). Однако у мужчин должна быть только одна копия рецессивного гена, сцепленного с Х-хромосомой, чтобы признак или расстройство проявились. Например, женщина может неосознанно нести рецессивный ген на одной из Х-хромосом и передать его сыну, который проявит эту черту или болезнь.
Каковы симптомы мышечной дистрофии?
Мышечная дистрофия обычно диагностируется у детей в возрасте от 3 до 6 лет.Ранние признаки заболевания включают задержку в ходьбе, трудности с подъемом из положения сидя или лежа и частое падение, при этом слабость, как правило, поражает плечо и тазовые мышцы в качестве одного из начальных симптомов. Ниже приведены наиболее частые симптомы мышечной дистрофии. Однако каждый ребенок может испытывать симптомы по-разному. Симптомы могут включать:
- Неуклюжие движения
- Трудности при подъеме по лестнице
- Часто спотыкаются и падают
- Невозможно нормально прыгать или прыгать
- Ходьба на цыпочках
- Боль в ногах
- Слабость лица
- Неспособность закрывать глаза или свистеть
- Слабость плеча и руки
Характерной клинической характеристикой мышечной дистрофии Дюшенна (МДД) является симптом Гауэрса.Детям с мышечной дистрофией Дюшенна очень трудно вставать из положения сидя или лежа на полу. Сначала они подтягиваются на четвереньки. Ребенок поднимает руки вверх по ногам, чтобы взять себя в руки при подъеме в положение стоя.
Кроме того, у детей с мышечной дистрофией часто бывают очень большие икры из-за большого количества жировых отложений, замещающих мышцы.
Симптомы мышечной дистрофии могут напоминать другие состояния или проблемы со здоровьем.Всегда консультируйтесь с врачом вашего ребенка для постановки диагноза.
Как диагностируется мышечная дистрофия?
Диагноз мышечной дистрофии ставится на основании медицинского осмотра и диагностического обследования лечащим врачом вашего ребенка. Во время обследования врач вашего ребенка собирает полную историю беременности и родов ребенка и спрашивает, есть ли у других членов семьи мышечная дистрофия.
Диагностические исследования мышечной дистрофии могут включать:
- Анализы крови.К ним относятся генетические анализы крови.
- Биопсия мышц. Первичный тест, используемый для подтверждения диагноза. Берется небольшой образец мышечной ткани и исследуется под микроскопом.
- Электромиограмма (ЭМГ). Тест, чтобы проверить, является ли мышечная слабость результатом разрушения мышечной ткани, а не повреждения нервов.
- Электрокардиограмма (ЭКГ или ЭКГ). Тест, который регистрирует электрическую активность сердца, выявляет аномальные ритмы (аритмии или аритмии) и обнаруживает повреждение сердечной мышцы.
 Первичный тест, используемый для подтверждения диагноза. Берется небольшой образец мышечной ткани и исследуется под микроскопом.
Первичный тест, используемый для подтверждения диагноза. Берется небольшой образец мышечной ткани и исследуется под микроскопом.Лечение мышечной дистрофии
Специфическое лечение мышечной дистрофии будет назначено врачом вашего ребенка на основании:
- Возраст вашего ребенка, общее состояние здоровья и история болезни
- Степень заболевания
- Тип состояния
- Толерантность вашего ребенка к конкретным лекарствам, процедурам или методам лечения
- Ожидания относительно течения состояния
- Ваше мнение или предпочтения
На сегодняшний день не существует известного лечения, лекарств или хирургии, которые вылечили бы мышечную дистрофию или остановить ослабление мышц.Цель лечения — предотвратить деформацию и дать ребенку возможность действовать максимально независимо.
Поскольку мышечная дистрофия — это пожизненное состояние, которое невозможно исправить, лечение включает в себя сосредоточение внимания на предотвращении или минимизации деформаций и максимальном повышении функциональных возможностей ребенка дома и в обществе.
Лечение мышечной дистрофии может быть нехирургическим или хирургическим. Нехирургические вмешательства могут включать:
- Физиотерапия
- Средства позиционирования, помогающие ребенку сидеть, лежать или стоять
- Подтяжки и шины, используемые для предотвращения деформации, поддержки или защиты
- Лекарства
- Консультации по питанию
- Психологическое консультирование
Хирургические вмешательства могут рассматриваться для лечения следующих состояний:
- Сколиоз (искривление позвоночника вбок), связанный с мышечной дистрофией
- Поддержание способности ребенка сидеть или стоять
Долгосрочная перспектива для ребенка с мышечной дистрофией
Мышечная дистрофия — это прогрессирующее заболевание, которое требует пожизненного лечения для предотвращения деформации и осложнений.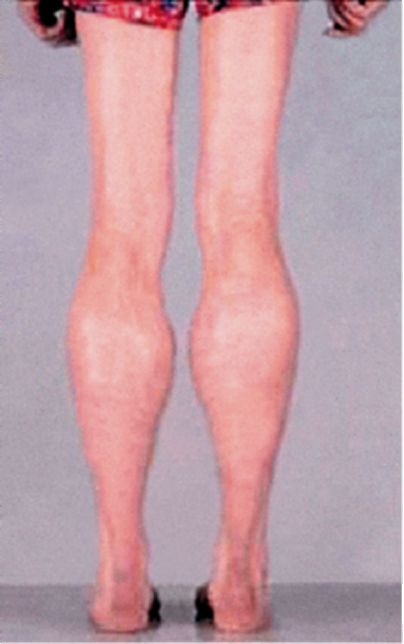 По мере роста ребенка часто становится труднее ходить и сидеть. Обычно к 12 годам ребенку нужна инвалидная коляска, потому что мышцы ног слишком слабы, чтобы работать. Проблемы с сердцем или легкими часто возникают к концу подросткового возраста или к началу 20-летнего возраста.
По мере роста ребенка часто становится труднее ходить и сидеть. Обычно к 12 годам ребенку нужна инвалидная коляска, потому что мышцы ног слишком слабы, чтобы работать. Проблемы с сердцем или легкими часто возникают к концу подросткового возраста или к началу 20-летнего возраста.
Междисциплинарная медицинская бригада будет работать с вашей семьей, чтобы улучшить функциональные результаты вашего ребенка и оказать поддержку, когда вы научитесь заботиться о потребностях вашего ребенка.
Ассоциация мышечной дистрофии может быть важным ресурсом, как в финансовом, так и в эмоциональном плане, для родителей детей с мышечной дистрофией.
Что такое мышечная дистрофия Дюшенна?
Что такое мышечная дистрофия Дюшенна?
Мышечная дистрофия Дюшенна — это генетическое заболевание, характеризующееся прогрессирующей потерей мышечной массы. Это мультисистемное заболевание, поражающее многие части тела и приводящее к ухудшению работы скелетных, сердечных и легких мышц.
Дюшенна вызвано изменением гена дистрофина. Без дистрофина мышцы не могут нормально функционировать или восстанавливать себя. Мышечный дистроф Беккера y, который менее серьезен, чем Дюшенна, возникает при производстве дистрофина, но не в нормальной форме или количестве.
Поскольку ген дистрофина находится на Х-хромосоме , он в первую очередь поражает мужчин, в то время как женщины обычно являются носителями. Однако у некоторых женщин могут проявляться различные физические симптомы Дюшенна, и поэтому они называются «проявляющими носителями».
Миссия и работа PPMD распространяется как на Дюшенна, так и на Беккера, но для простоты мы в первую очередь ссылаемся на Дюшенна.
Что вызывает Дюшенна?
Дюшенна вызван мутацией в гене, который кодирует дистрофин, белок, который необходим для правильного функционирования наших мышц. Без дистрофина мышцы не могут нормально функционировать или восстанавливать себя. Затем потеря мышц приводит к потере силы и функций.
Затем потеря мышц приводит к потере силы и функций.
Дюшенна может передаваться от родителей к ребенку или быть результатом случайных спонтанных генетических мутаций, которые могут произойти во время любой беременности.Фактически, около случаев заболевания Дюшенна из каждых трех случаев происходят в семьях, ранее не имевших заболевания Дюшенна. Другими словами, он может повлиять на кого угодно и распространяется на все расы и культуры. Узнайте больше о том, что вызывает болезнь Дюшенна и Беккера и как это может повлиять на более чем одного человека в семье.
Узнайте о генетических причинахКаковы признаки и симптомы Дюшенна?
Средний возраст диагноза Дюшенна составляет около 4 лет. Часто будет задержка на ранних этапах развития, таких как сидение, ходьба и / или разговор.Задержка речи и / или неспособность идти в ногу со сверстниками часто являются первыми признаками расстройства. Симптомы Беккера могут начаться в детстве, подростковом возрасте или даже позже. Узнайте больше о признаках и симптомах Дюшенна и Беккера.
Узнайте о признаках и симптомахКаковы успехи Дюшена?
Дюшенн прогрессирует по-разному для каждого человека. Даже у братьев и сестер с одной и той же мутацией симптомы могут сильно отличаться.
Потеря мышечной массы впервые замечается в детстве, когда происходит потеря силы, функции и гибкости бедер, бедер, плеч и таза.В подростковом возрасте эти потери начинают распространяться на руки, голени и туловище. Поскольку также отсутствует дистрофин в мышцах сердца и легких, это также влияет на работу сердца и дыхание. Кроме того, у некоторых людей могут быть проблемы с обучением и поведением из-за нехватки дистрофина в головном мозге.
Симптомы Дюшенна прогрессируют по спектру от позднего начала / очень легких симптомов до раннего начала / тяжелых симптомов. Регулярные посещения вашей нервно-мышечной бригады помогут вам отслеживать прогрессирование этого заболевания и способы его лечения на этом пути. Узнайте больше о прогрессе Дюшенна.
Узнайте больше о прогрессе Дюшенна.
Какова средняя продолжительность жизни Дюшенна?
При улучшении ухода за больными Дюшенна все больше людей доживают до 30 лет и старше. Поскольку клиническая помощь продолжает улучшаться, а на горизонте ожидаются клинические испытания, исследования и методы лечения, мы надеемся улучшить качество и количество жизни с Дюшеном до гораздо более старшего возраста.
Почему важно знать конкретную мутацию Дюшенна?
Многие методы лечения Дюшенна, разрабатываемые и / или одобренные для лечения Дюшенна, являются «специфичными для мутаций», что означает, что они принесут пользу только людям с определенными мутациями.Вы должны знать свою мутацию, чтобы участвовать в клинических испытаниях и получить доступ к любым текущим или будущим методам лечения, специфичным для мутаций. Если вы никогда не проходили генетическое тестирование или вам необходимо повторное генетическое тестирование, PPMD может предоставить бесплатное генетическое тестирование подходящим людям через нашу программу Decode Duchenne.
Узнайте о типах мутацийЧто значит быть перевозчиком?
Носитель — женщина, у которой есть мутация в одной из двух копий гена дистрофина.У носителей больше шансов иметь сыновей от Дюшенна и дочерей, которые являются носителями.
Самки-носители обычно не подвержены влиянию Дюшенна или Беккера, потому что они вырабатывают достаточно белка дистрофина. Однако у них могут быть некоторые симптомы Дюшенна, такие как мышечная слабость и проблемы с сердцем. Хотя это случается редко, у некоторых женщин могут быть классические симптомы Дюшенна. Все носители должны быть проверены врачом, знакомым с Дюшенном. Узнайте больше о том, что значит быть перевозчиком.
Узнайте о перевозчикахЧто делать, если вы не уверены, что у вашего ребенка Дюшенн?
Постановка точного и своевременного диагноза является важным аспектом лечения. Есть несколько инструментов, которые вы и ваш лечащий врач можете использовать для оценки вашего ребенка:
Узнайте больше о признаках Дюшенна и о том, как его диагностировать.
Каков текущий статус ухода и лечения Дюшенна?
Хотя в настоящее время нет лекарства от Дюшенна, надежда есть — возможно, больше, чем когда-либо прежде.PPMD находится в авангарде достижений в области ухода и лечения Дюшенна. Мы применяем передовой подход, чтобы ускорить поиск методов лечения, которые положат конец Дюшенну, для каждого человека, затронутого этой болезнью.
Улучшения в уходе
Мы стали свидетелями величайших достижений в борьбе за искоренение Дюшенна в отношении стандартов медицинской помощи, что привело к значительному увеличению количества и качества жизни. Благодаря стремлению PPMD продвигать уход за больными, люди с Дюшенном живут дольше, сильнее, счастливее и продуктивнее.Члены нашего сообщества занимают важную работу, влияют на политику в Вашингтоне, женятся и заводят семьи — то, что мы не могли представить себе даже 10 лет назад.
Успехи в исследованиях и лечении
PPMD инвестировала более 50 миллионов долларов в инновационные исследования, которые повлияют на все заболевание, независимо от мутации, возраста или стадии прогрессирования, включая инвестиции в стратегии и методы лечения, восстанавливающие или заменяющие дистрофин (например, генная терапия, пропуск экзона, CRISPR и т. д.), а также те, которые лечат симптомы (например, те, которые нацелены на фиброз, воспаление, кровоток и т. д.), зная, что лечение Дюшенна, вероятно, будет включать комбинацию терапий.
С 2016 года FDA одобрило два лечения в Дюшенне с дополнительными видами лечения, приближающимися к нормативной финишной черте:
- Эмфлаза — кортикостероид, обладающий противовоспалительным и иммунодепрессивным действием.
- EXONDYS 51 представляет собой антисмысловой олигонуклеотид, указанный для лечения Дюшенна у пациентов с подтвержденной мутацией гена Дюшенна, которая поддается пропуску экзона 51.
Мышечная дистрофия | Институт Кеннеди Кригера
Ниже описаны некоторые из распространенных типов мышечных дистрофий. В Kennedy Krieger мы имеем опыт и приветствуем людей с редкими и распространенными мышечными заболеваниями.
Мышечная дистрофия Дюшенна:
Мышечная дистрофия Дюшенна — одно из наиболее распространенных наследственных заболеваний во всем мире. Это заболевание поражает почти исключительно мальчиков. Родители могут сначала увидеть, что их ребенок от трех до пяти лет часто падает, медленно бегает, ходит на цыпочках или ходит вразвалочку.Икры у ребенка часто бывают необычно большими. Первоначально слабость наиболее выражена в мышцах бедер и верхней части ног, но со временем она будет распространяться на большинство произвольных мышц, включая те, которые отвечают за дыхание. Сердце тоже со временем слабеет. Из-за слабости сердца и дыхательных мышц это заболевание становится смертельным и требует тщательного медицинского лечения.
Подробнее о мышечной дистрофии Дюшенна
Facioscapulohumeral Muscular Dystrophy:
Лицево-плечевой (ФСГ) дистрофия — распространенная мышечная дистрофия, при которой наблюдается прогрессирующая слабость лица, плеч и плеч, а также ног.Симптомы дистрофии ФСГ могут проявляться в детстве с сильной слабостью лица и конечностей или медленно и постепенно развиваться во взрослом возрасте с прогрессирующими трудностями при закрытии глаз и поднятии предметов, а также при спотыкании. Заболевание вызвано дегенерацией мышц из-за определенной делеции хромосомы. Это удаление передается от одного поколения к другому.
Узнать больше о фациоскапуло-плечевой дистрофии
Миотоническая мышечная дистрофия:
Миотоническая мышечная дистрофия — распространенное мультисистемное заболевание, которое поражает скелетные мышцы (мышцы, двигающие конечности и туловище), а также гладкие мышцы (мышцы, контролирующие пищеварительную систему) и сердечные мышцы сердца.Симптомы миотонической дистрофии могут включать затруднения при отпускании хватки (миотония), слабость мышц рук и ног, затрудненное глотание и нарушение сердечного ритма.