
Сахарный диабет врожденный: Клинический случай неонатального сахарного диабета, обусловленного мутацией гена INS | Атанесян
Клинический случай неонатального сахарного диабета, обусловленного мутацией гена INS | Атанесян
Неонатальный сахарный диабет (НСД) – серьезная патология эндокринной системы, диагностируемая у детей первых 6 мес жизни. НСД является редким (1 : 300 000–1 : 400 000 новорожденных) заболеванием, обусловленным панкреатической β-клеточной дисфункцией, проявляющейся дефицитом инсулина и симптомами гипергликемии [1, 2].
Благодаря широкому внедрению молекулярной диагностики представление об этиологии и патогенезе неонатального диабета кардинальным образом изменилось. В относительно недавнем времени НСД рассматривали как вариант диабета 1-го типа, манифестирующего в раннем возрасте. Однако проведенные в течение последних десятилетий исследования продемонстрировали, что НСД является не аутоиммунным заболеванием, а результатом множественных мутаций в различных генах, кодирующих белки, выполняющие важную роль в нормальном функционировании β-клеток островков Лангерганса поджелудочной железы [3, 4].
Очевидно на сегодняшний день, что результаты молекулярно-генетической диагностики различных форм НСД оказывают влияние на выбор терапии заболевания и определяют его прогноз. Это особенно актуально для пациентов, имеющих мутации в генах KCNJ11 или АВСС8, кодирующих две белковые субъединицы (Kir6.2 и SUR1 соответственно) АТФ-чувствительных калиевых каналов [1, 3, 5, 6].
В основе этиологии НСД лежит патология участка хромосомы 6q24 и активация мутации в генах, кодирующих гликолитические ферменты, глюкокиназу и две белковые субъединицы (Kir6.2 и SUR1) панкреатического АТФ-канала β-клеток поджелудочной железы [3, 7, 8]. Выявлены мутации ряда генов (IPF1, PTF1A, FOXP3, E1F2AK3, HNF1B, GLIS3), которые могут вызывать мультисистемные заболевания, включая НСД [9, 10]. Определение аномалий в структуре 6-й хромосомы, мутаций в KCNJ11 и ABCC8 генах позволяет проводить дифференциальную диагностику между транзиторной и перманентной формами НСД уже в периоде новорожденности [1, 6, 7].
В настоящее время определена мутация гена инсулина (INS), которая является также одной из причин неонатального диабета, при этом с генетической точки зрения важно, что большинство выявленных мутаций не наследуются, а являются спорадическими [5, 9, 11].
Аутоиммунный СД 1 типа крайне редко манифестирует у детей младше шестимесячного возраста. Существуют специфические подтипы моногенного СД, которые могут иногда встречаться у младенцев раннего возраста, однако сейчас считается, что большинство таких случаев связано с мутациями FOX3, а не с СД 1 типа [11, 12]. Таким образом, все пациенты, диагноз которым был установлен до шестимесячного возраста, нуждаются в генетическом обследовании с целью исключения моногенной формы НСД.
Большинство пациентов с НСД рождаются с дефицитом массы тела, отражающим пренатальный дефицит секреции инсулина, поскольку в антенатальном периоде инсулин является важнейшим ростовым фактором для плода.
Пациентам с НСД в половине случаев требуется лечение в течение всей жизни, так как у них чаще диагностируется перманентная форма неонатального диабета. В остальных случаях НСД переходит в стадию ремиссии в течение нескольких недель или месяцев – это транзиторный неонатальный диабет, однако он может рецидивировать в последующем [12]. НСД в основном проявляет себя как изолированное нарушение, однако у некоторых пациентов присутствуют различные внепанкреатические клинические признаки, связанные с определенным геном [2, 3].
В остальных случаях НСД переходит в стадию ремиссии в течение нескольких недель или месяцев – это транзиторный неонатальный диабет, однако он может рецидивировать в последующем [12]. НСД в основном проявляет себя как изолированное нарушение, однако у некоторых пациентов присутствуют различные внепанкреатические клинические признаки, связанные с определенным геном [2, 3].
Транзиторный НСД манифестирует вскоре после рождения, а затем подвергается спонтанной ремиссии во время младенчества, но может рецидивировать и переходить в постоянную форму СД в детстве или юности [11]. Генетическая основа транзиторного НСД примерно в 70% случаев представлена аберрациями в хромосоме 6q24, которые вызывают избыточную экспрессию двух импринтированных генов PLAGL1 и HYMAI, тогда как большинство остальных случаев связано с активирующими мутациями одного из двух генов, кодирующих две субъединицы АТФ-чувствительного калиевого канала на мембране β-клетки (KCNJ11 или ABCC8) [13]. Небольшое количество случаев транзиторного НСД связано с мутациями других генов, в том числе HNF1B, INS (ген препроинсулина) и др. [6, 11].
Небольшое количество случаев транзиторного НСД связано с мутациями других генов, в том числе HNF1B, INS (ген препроинсулина) и др. [6, 11].
Транзиторный НСД, обусловленный аномалиями импринтинга хромосомы 6q24, можно отличить от других типов неонатального диабета по весу ребенка при рождении, наличию пороков развития, указывающих на этиологическую подгруппу. У пациентов с транзиторным НСД, вызванным мутациями генов АТФ-чувствительных калиевых каналов, ремиссия диабета обычно наступает позже, а последующий рецидив раньше, чем у больных транзиторным НСД, вызванным мутациями 6q24 [7]. Для понимания генотипически-фенотипических особенностей неонатального диабета крайне важна молекулярно-генетическая диагностика, которая позволяет не только верифицировать мутацию, но и в значительной степени предсказать прогноз заболевания [4, 8].
Наиболее частыми причинами перманентного НСД являются мутации KCNJ11 (приблизительно в 30–50% случаев), ABCC8 (9–10%) и INS (12–20%) [1, 14, 15]. В 20–30% случаев этиология перманентного НСД остается неизвестной, хотя самой распространенной его причиной в аутбредных популяциях являются мутации генов АТФ-чувствительного калиевого канала или INS [3]. В случае близкородственных браков самая распространенная этиология – синдром Уолкотта–Раллисона, или гомозиготные мутации гена GCK [14].
В 20–30% случаев этиология перманентного НСД остается неизвестной, хотя самой распространенной его причиной в аутбредных популяциях являются мутации генов АТФ-чувствительного калиевого канала или INS [3]. В случае близкородственных браков самая распространенная этиология – синдром Уолкотта–Раллисона, или гомозиготные мутации гена GCK [14].
АТФ-чувствительные калиевые каналы – это гетерооктамерные комплексы, состоящие из четырех порообразующих 2-субъединиц и четырех регулирующих SUR1-субъединиц, кодируемых генами KCNJ11 и ABCC8 соответственно. Они регулируют секрецию инсулина, связывая внутриклеточный метаболизм с электрической активностью β-клеток. Повышение внутриклеточной метаболической активности вызывает изменения в соотношении АТФ/АДФ в панкреатических β-клетках, вследствие чего закрываются АТФ-чувствительные калиевые каналы, происходит деполяризация клеточной мембраны, а в результате этого процесса инициируется секреция инсулина (рис. 1) [13].
Рис. 1. Регуляция секреции инсулина β-клетками поджелудочной железы: SUR – рецептор сульфонилмочевины; ПСМ – препарат сульфонилмочевины; АТФ – аденозинтрифосфат; АДФ – аденозиндифосфат.
Активирующие мутации генов KCNJ11 и ABCC8 не позволяют каналам закрываться, тем самым предотвращая секрецию инсулина в ответ на гипергликемию, и являются самой распространенной причиной перманентного НСД, а также второй по частоте причиной транзиторного НСД [6]. У большинства пациентов с мутациями KCNJ11 диабет перманентный, а не транзиторный. И наоборот, мутации ABCC8 чаще, примерно в 66% случаев, вызывают транзиторный НСД [13].
У пациентов с мутациями в генах АТФ-чувствительных калиевых каналов клиническая картина заболевания представлена инсулинозависимостью с низкими или нулевыми показателями С-пептида, при этом характерна манифестация заболевания с диабетического кетоацидоза. В 20% случаев, обусловленных мутациями гена KCNJ11, манифестация заболевания, помимо диабета, часто включает неврологические проявления ввиду экспрессии генов АТФ-чувствительных каналов в нейронах и мышечных клетках [11].
Активирующие мутации KCNJ11, вызывающие НСД, всегда гетерозиготны. Так как примерно в 90% случаях эти мутации возникают de novo, в семейном анамнезе неонатальный диабет обычно отсутствует, однако в случаях наследственного диабета рассматривается аутосомно-доминантный тип наследования. Риск возникновения диабета у детей, рожденных пациентками с НСД, равен 50%, что характерно для большинства больных, имеющих активирующие мутации ABCC8. Неонатальный диабет у них наследуется рецессивно в тех случаях, когда пациенты гомозиготны или компаунд-гетерозиготны по двум разным мутациям [3].
Второй по распространенности причиной перманентного НСД после мутаций генов натрий-калиевых АТФ-каналов являются гетерозиготные кодирующие мутации гена INS [6]. Доминирующая часть гетерозиготных мутаций INS являются спорадическими мутациями de novo, и лишь 20% пробандов имеют аутосомно-доминантный неонатальный диабет в генеалогическом анамнезе [1, 16]. Наряду с гетерозиготными мутациями INS существует информация о гомозиготных или компаунд-гетерозиготных мутациях, вызывающих НСД [3, 13].
Мутации в гене инсулина могут нарушать свертывание его белка-предшественника и проинсулина, заменяя остаток цистеина, который необходим для образования дисульфидной связи, тем самым блокируя образование инсулина. «Мутантный» инсулин образует метастабильное состояние с большими эффектами на его N-концевую область, что включает в себя образование частично сложенного промежуточного продукта с конформационным изменением в N-концевой области цепи A, которая генерирует гибкий N-концевой домен [17]. Это может привести к аномальным взаимодействиям с другими проинсулинами в процессе агрегации. Агрегация белков сопровождается частично сложенным промежуточным продуктом с изменениями вторичной структуры, такими как потеря вторичной структуры α-спирали. Увеличение количества неправильных дисульфидных связей в белковых комплексах проявляется доминантным отрицательным эффектом с присутствием неспаренного цистеина в проинсулине. Эти несогласованные белки сохраняются в эндоплазматическом ретикулуме (ЭР) β-клетки, ингибируя нормальную функцию и, вероятно, приводя к гибели β-клеток через ЭР-стресс [17, 18].
Пациенты с гетерозиготными мутациями гена INS характеризуются тяжелой задержкой внутриутробного развития, как и пациенты с мутациями генов калиевых каналов в панкреатических клетках. Однако клиническая симптоматика диабета дебютирует в более позднем возрасте, хотя сроки могут и пересекаться, а неврологические симптомы у пациентов не манифестируют, что является прямым следствием мутации.
Иногда мутации гена INS вызывают перманентный НСД после шестимесячного возраста ребенка, и поэтому в некоторых ситуациях показано генетическое обследование, особенно пациентам с СД 1 типа при отсутствии антител [19].
Генетическое обследование позволяет определиться с тактикой терапии, так, пациенты с активирующими мутациями генов АТФ-чувствительного калиевого канала могут быть переведены на препараты группы сульфонилмочевины, что является вполне патофизиологически аргументированным подходом к лечению. Пероральная терапия препаратами сульфонилмочевины безопасна и эффективна в краткосрочной перспективе у большинства пациентов с диабетом, связанным с мутацией SUR1, и может успешно заменить лечение инсулиновыми инъекциями [18].
НСД, обусловленный мутациями гена INS, как правило, требует использования инсулина. Раннее начало терапии инсулином, вероятно, снижает потребность в эндогенном производстве инсулина β-клетками поджелудочной железы, сокращая тем самым количество несогласованного проинсулина, который вырабатывается и сохраняется в ЭР. Этот эффект может позволить уменьшить гибель β-клеток и сохранить некоторое количество эндогенного производства инсулина и общую β-клеточную функцию [18, 20].
Таким образом, результаты генетического обследования не только необходимы для дифференциальной диагностики формы заболевания, но обеспечивают персонализированный подход к терапии пациентов с НСД.
Большинство научных исследований этой крайне редкой патологии основаны на анализе единичных случаев, поэтому мы также сочли возможным привести собственное клиническое наблюдение девочки с НСД, впервые диагностированным на 3-м месяце жизни.
ОПИСАНИЕ СЛУЧАЯ
Пациентка С., в возрасте 8 мес впервые оказалась на приеме у детского эндокринолога с жалобами на частые гипогликемии до 3–4 раз/месяц, суточные колебания глюкозы крови и частые гипергликемии до 24,0 ммоль/л.
Анамнез жизни: девочка от 2-й беременности (1-я беременность закончилась самопроизвольным абортом). Беременность протекала с осложнениями (в I триместре – угроза прерывания, в III триместре – фето-плацентарная недостаточность). Роды первые, срочные. Масса при рождении составила 2450 г, длина – 49 см, оценка по шкале Апгар 8/9 баллов.
Ребенок выписан из родильного дома на 5-е сутки в удовлетворительном состоянии. Находилась на смешанном вскармливании до 2-недельного возраста, затем вскармливание искусственное. Прорезывание зубов с 7 мес. Нервно-психическое развитие без особенностей: ребенок держит голову с 1,5–2 мес, сидит с 6–7 мес.
Родители не состоят в кровном родстве, русские по национальности. Матери к моменту рождения ребенка было 24 года, отцу – 29 лет. Со стороны матери – у бабушки артериальная гипертензия, хроническая варикозная болезнь нижних конечностей, у матери пациентки врожденная тромбофилия, со стороны отца – у бабушки гипертоническая болезнь, отец девочки здоров.
Со слов мамы, в возрасте 3 мес стала отмечать полифагию, отсутствие адекватной прибавки в весе, беспокойство в течение всего дня. При обследовании в возрасте 5 мес у педиатра по месту жительства диагностированы: в ОАМ – глюкозурия, кетонурия (+++), глюкоза крови – 20,0 ммоль/л. Ребенок был госпитализирован в ОРИТ по поводу кетоацидоза в ДГКБ им. Г.К. Филиппского г. Ставрополя. В стационаре был установлен диагноз «Сахарный диабет, 1 типа», инициирована инсулинотерапия (инсулин Лизпро (Хумалог) 0,5–1,0 Ед перед приемами пищи при глюкозе крови выше 10,0 ммоль/л + Гларгин (Лантус) 2 Ед/сут в 20.00), девочка выписана под наблюдение эндокринолога по месту жительства.
С возраста 5 мес и до 8 мес девочка наблюдалась эндокринологом по месту жительства (результаты гликированного гемоглобина – 12,3%), проводилась коррекция дозы продленного инсулина (увеличена доза Гларгина до 3 ЕД/сут).
В возрасте 8 мес девочка направлена на госпитализацию в педиатрическое отделение НМИЦ им. В.А. Алмазова (Санкт-Петербург) с целью уточнения этиологии СД. В отделении было проведено молекулярно-генетическое обследование. При поступлении ребенок находился на интенсифицированной инсулинотерапии (Лизпро 0,5 Ед при показателях глюкозы крови выше 10,0 ммоль/л и 2–3 Ед/сут Гларгина).
Алмазова (Санкт-Петербург) с целью уточнения этиологии СД. В отделении было проведено молекулярно-генетическое обследование. При поступлении ребенок находился на интенсифицированной инсулинотерапии (Лизпро 0,5 Ед при показателях глюкозы крови выше 10,0 ммоль/л и 2–3 Ед/сут Гларгина).
Результаты физикального и лабораторно-инструментального исследования
Объективный статус на момент осмотра: общее состояние удовлетворительное. Сознание ясное. Рост – 68 см (-0,3 SDS), вес – 6,8 кг (-1,5 SDS), ИМТ – 14,78 кг/м
Тоны сердца ясные, ритмичные, ЧСС – 117 в минуту. Аускультативно дыхание в легких везикулярное. Живот не увеличен, мягкий при пальпации. Размеры печени и селезенки пальпаторно не увеличены.
Наружные половые органы развиты правильно по женскому типу. Вторичные половые признаки допубертатные (стадия по Таннеру 1).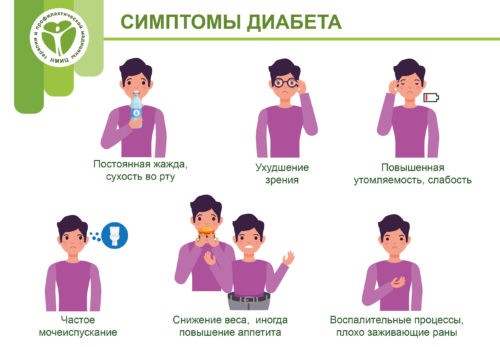
Лабораторное обследование
ОАК: WBC – 8,0*109/л, HGB – 104 г/л, RBC – 3,5*1012/л, PLT – 245*109/л, СОЭ – 65 мм/ч, нейтрофилы – 14,5%, лимфоциты – 71,6%, моноциты – 11,5%, эозинофилы – 2,3%, базофилы – 0,1%.
ОАМ – прозрачная, удельный вес – 1.041, кислотность – 5,5 ед. pH, глюкоза >56 ммоль/л, кетоновые тела – отр., белок, нитриты, эритроциты, лейкоциты, уробилиноген – не определяются.
Биохимический анализ крови: общий белок – 66,0 г/л, натрий – 136 ммоль/л, калий – 4,4 ммоль/л, билирубин общий – 4,0 мкмоль/л, холестерин общий – 4,07 ммоль/л, триглицериды – 1,17 ммоль/л, холестерин ЛПВП – 1,13 ммоль/л, холестерин ЛПОНП – 0,54 ммоль/л, КА – 2,6, креатинин – 43,0 мкмоль/л, АЛТ – 18,0 Ед/л, АСТ – 32,0 Ед/л, фосфор – 1,66 ммоль/л.
Гликированный гемоглобин – 9,1%.
ТТГ – 1,61 мМе/л, свободный Т4 – 12,6 пмоль/л.
В таблице 1 представлены показатели гликемического профиля пациентки при поступлении в стационар (до коррекции терапии).
Таблица 1. Гликемический профиль пациентки С. при поступлении в стационар
08.00 | 11.30 | 13.30 | 15.30 | 17.30 | 19.30 | 22.00 | 01.00 | 04.00 |
11,7 | 16,6 | 14,1 | 11,5 | 15,8 | 16,8 | 11,1 | 4,2 | 12,2 |
11,0 | 12,3 | 11,0 | 3,9 | 14,05 | 10,02 | 8,5 | 6,7 | 13,1 |
12,16 | 14,2 | 10,6 | 10,3 | 17,1 | 10,8 | — | 13,3 | 11,6 |
11,8 | 9,3 | 9,7 | 9,8 | 10,2 | 11,7 | 8,2 | 11,8 | 7,5 |
Инструментальное обследование
ЭКГ: ритм синусовый с ЧСС 129 в минуту. Положение электрической оси сердца: нормальное. По форме ЭКГ без патологии.
Положение электрической оси сердца: нормальное. По форме ЭКГ без патологии.
УЗИ печени, поджелудочной железы, желчного пузыря, почек, надпочечников: Эхо-признаки умеренной гепатомегалии, функциональная деформация желчного пузыря, минимальная каликоэктазия правой почки.
УЗИ щитовидной железы: без патологии.
КТ головного мозга: умеренная наружная неокклюзионная гидроцефалия.
Консультация невролога: перинатальная энцефалопатия постгипоксического генеза, поздний восстановительный период. Синдром двигательных нарушений, мышечная гипотония, улучшение.
Консультация нейрохирурга: в нейрохирургическом лечении не нуждается.
Результаты генетического обследования (метод параллельного секвенирования – платформа lon Torrent): по данным генетического обследования: в гене INS выявлен гетерозиготный вариант с. 125Т> G p.V42G, чаще ассоциированный с перманентным НСД.
Учитывая лабильность течения СД, состояние декомпенсации, а также с целью улучшения качества жизни ребенка в раннем возрасте в отделении принято решение об установке инсулиновой помпы. В отделении проведен подбор скорости инсулина под контролем суточной гликемии, мама обучена технике пользования инсулиновой помпой.
В отделении проведен подбор скорости инсулина под контролем суточной гликемии, мама обучена технике пользования инсулиновой помпой.
Ребенок переведен на ПИТ (система введения инсулина Accu-Chek Combo). Базальная доза составила: 08:00–01:00 – 0,11 Ед/ч, 01:00–08:00 – 0,08 Ед/ч. Углеводные коэффициенты: на завтрак – 1 ХЕ=0,7 Ед; обед – 1 ХЕ=0,6 Ед; ужин – 1 ХЕ=0,5 Ед. Коэффициент чувствительности: 1 Ед – 15–20 ммоль/л. Результаты гликемического профиля пациентки после перевода на помповую инсулинотерапию представлены в табл. 2
Таблица 2. Гликемический профиль пациентки С. на фоне перевода на помповую инсулинотерапию
08.00 | 11.30 | 13.30 | 15.30 | 17.30 | 19.30 | 22.00 | 01.00 | 04. |
5,8 | 7,6 | 7,7 | 7,5 | 10,2 | 4,0 | 8,4 | 8,7 | 9,5 |
7,12 | 6,93 | 4,3 | 8,1 | 11,2 | 8,51 | 8,84 | 8,2 | 7,6 |
6,99 | 10,3 | 8,11 | 7,5 | 6,8 | 8,17 | 7,5 | 9,5 | 7,7 |
10,1 | 8,12 | 6,7 | 8,16 | 12,6 | 7,88 | 5,5 | 6,8 | 9,3 |
 00
00ОБСУЖДЕНИЕ
Таким образом, учитывая раннюю манифестацию СД (до шестимесячного возраста), а также результаты проведенного генетического обследования, диагноз НСД не вызывает сомнений.
Пациентке с целью купирования кетоацидоза и нормализации показателей глюкозы крови на старте назначена терапия инсулином. Предполагалась коррекция лечения после получения результатов генетического обследования. Благодаря возможности выполнения генетической диагностики, была выявлена мутация гена INS, которая чаще ассоциирована с перманентной формой НСД, что позволило определиться с тактикой лечения в пользу продолжения инсулинотерапии.
Очевидно, что модель детского питания является непредсказуемой и сложной, что связано с различной продолжительностью и объемом кормления, сложностью подсчета углеводов, в свою очередь, обусловливает высокую вероятность гипогликемических состояний у детей грудного возраста. Несомненно, дети раннего возраста нуждаются в назначении инсулинотерапии с помощью инсулиновой помпы с целью достижения компенсации заболевания, профилактики микро- и макрососудистых осложнений, повышения качества жизни и, конечно, безопасного и эффективного управления диабетом. Результаты гликемического профиля девочки, выполненного после установления инсулиновой помпы, ярко демонстрируют улучшения показателей углеводного обмена, снижение суточной вариабельности гликемии и частоту гипогликемий.
Результаты гликемического профиля девочки, выполненного после установления инсулиновой помпы, ярко демонстрируют улучшения показателей углеводного обмена, снижение суточной вариабельности гликемии и частоту гипогликемий.
ЗАКЛЮЧЕНИЕ
В настоящее время не существует данных, которые позволили бы составить фенотипический и генотипический «портрет» форм НСД, а также уточнить факторы, определяющие их возникновение. Требуется дальнейшее изучение клинических случаев НСД с целью определения характеристики его подтипов с последующим прогнозированием заболевания
Результаты молекулярно-генетической диагностики, благодаря которой появилась возможность верифицировать мутацию, позволяют проводить дифференциальную диагностику между транзиторной и перманентной формами НСД уже в периоде новорожденности, что, бесспорно, важно для выбора тактики лечения и ведения пациента в будущем.
Выполнение генетического тестирования не только является неотъемлемым этапом диагностики НСД, но и основой выбора персонифицированной терапии, так как мутации KCNJ11 и ABCC8 чувствительны к препаратам сульфонилмочевины, в то время как пациенты с мутациями гена INS, как правило, должны продолжать инсулинотерапию.
Предполагается, что раннее начало инсулинотерапии может привести к сохранению функции β-клеток, снижению дозы инсулина и улучшению гликемического контроля, что потенциально уменьшит риск осложнений, связанных с диабетом в последующем.
Очевидно, крайне важным является медико-генетическое консультирование семьи ребенка с НСД, генетическое тестирование детей, рожденных в данных семьях в последующем, что не только позволит своевременно диагностировать диабет, прогнозировать клиническое течение, но и снизить риск развития диабетического кетоацидоза в дебюте заболевания.
ДОПОЛНИТЕЛЬНАЯ ИНФОРМАЦИЯ
Источник финансирования. Молекулярно-генетическое исследование было проведено за счет бюджетных средств в НМИЦ им. В.А. Алмазова.
Согласие пациента. Родители пациентки добровольно подписали информированное согласие на публикацию персональной медицинской информации в обезличенной форме в журнале «Сахарный диабет».
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Участие авторов. Климов Л.Я., Атанесян Р.А., Углова Т.А., Вдовина Т.М., Долбня С.В. – концепция и дизайн; Костанова М.Ю., Стоян М.В., Курьянинова В.А., Алавердян Л.С., Долбня С.В. – сбор и обработка материала; Атанесян Р.А. – написание текста; Климов Л.Я. – редактирование. Все авторы внесли существенный вклад в проведение исследования и подготовку статьи, прочли и одобрили финальную версию перед публикацией.
Неонатальный сахарный диабет | ФГБУ «НМИЦ эндокринологии» Минздрава России
Неонатальный сахарный диабет (НСД) — это сахарный диабет, который был диагностирован в течение первых 6 месяцев жизни ребенка. Возраст начала диабета имеет принципиальное значение, т.к. только диабет развившийся именно до 6 месяца жизни является основным показанием для проведения ребенку молекулярно-генетического исследования.
Основные типы НСД
Транзиторный НСД чаще возникает в течение первых недель, а иногда и дней жизни ребенка. Характерны задержка внутриутробного развития, высокий уровень гликемии и значительная потребность в инсулинотерапии.
Характерны задержка внутриутробного развития, высокий уровень гликемии и значительная потребность в инсулинотерапии.
В возрасте 3-18 месяцев развивается ремиссия (т. е. исчезновение симптомов) заболевания, однако в старшем возрасте возможен рецидив СД,поэтому даже в случае отмены инсулинотерапии ребенок продолжает наблюдаться эндокринологом.
При перманентном (постоянном) НСД сахароснижающая терапия необходима на протяжении всей жизни ребенка. Реже встречаются синдромальные формы НСД, т.е. сочетание НСД с поражением других органов или систем
Когда можно заподозрить НСД?
В отличие от детей старшего возраста у которых первыми признаками СД являются жажда и частое обильное мочеиспускание (полиурия), наиболее постоянным признаком заболевания у грудных детей является отсутствие прибавки веса или потеря массы тела при адекватном режиме кормления ребенка. Реже родители отмечают появление «крахмальных» пятен на пеленках, липкой мочи, рецидивирующее течение дерматита в паховой области. Иногда НСД выявляется при диспансерном обследовании, например перед вакцинацией, или при сдаче анализов на фоне сопутствующих заболеваний.
Причины развития НСД?
НСД возникает в результате дефекта (мутаций) одного из генов, которые отвечают за нормальное развитие поджелудочной железы, синтез и работу инсулина. Заболевание может передаваться по наследству или возникает спонтанно (т.е. ребенок с НСД может родиться у родителей, не болеющих СД).При этом наиболее частыми причинами перманентного НСД являются мутации в генах KCNJ11, АВСС8 и в гене инсулина (INS). Нужно отметить, что у 25-30% пациентов с дефектами генов KCNJ11 и АВСС8 помимо НСД встречаются неврологические нарушения (эпилепсия, задержка психомоторного и речевого развития) – DEND или iDEND-синдром. Большинство случаев ТНСД связано с аномалиями хромосомы 6q24, реже встречаются дефекты генов KCNJ11, АВВС8 и INS.
Обязательно ли проведение молекулярно-генетического исследования ребенку с НСД?
Если ребенок заболел СД в первые 6 месяцев жизни, проведение генетического исследования обязательно, т. к. при выявлении мутаций в генах KCNJ11 или АВСС8 в большинстве случаев возможно назначение таблетированных сахароснижающих препаратов.
к. при выявлении мутаций в генах KCNJ11 или АВСС8 в большинстве случаев возможно назначение таблетированных сахароснижающих препаратов.
Нужно ли проводить генетическое исследование ребенку, заболевшему СД в 7-12 месяцев жизни, при отсутствии других критериев моногенного СД?
В возрастной группе 7-12 месяцев преимущественно встречается аутоиммунный СД1 типа, генетически обусловленные формы СД выявляются редко. Поэтому детям с дебютом СД от 7 до 12 месяцев жизни предварительно проводится исследование аутоиммунных маркеров СД1 типа. Если антитела отрицательные, решается вопрос о проведении молекулярно-генетического исследования (особенно при отягощенной наследственности по СД). Если антитела положительные, диагностируется СД1 типа и генетическое исследование не проводится.
Врожденный сахарный диабет — PubMed
Обзор
. 2020 авг; 72 (4): 240-249.
doi: 10.
Дарио Яфуско 1 , Анджела Занфардино 2 , Риккардо Бонфанти 3 , Ивана Раббоне 4 , Надя Тинто 5 , Фернанда Яфуско 5 , Серена Меола 5 , Мария Ф. Гиккино 2 , Гульсум Озен 6 , Франческа Касабуро 2 , Алессия Пископо 2 , Эмануэле Миралья-дель-Джудиче 2 , Фабрицио Барбетти 7
Принадлежности
- 1 Кафедра педиатрии Университета Кампании «Луиджи Ванвителли», Неаполь, Италия – dario.
 [email protected].
[email protected]. - 2 Кафедра педиатрии Кампанского университета им. Луиджи Ванвителли, Неаполь, Италия.
- 3 Отделение детской диабетологии, Отделение педиатрии, Научно-исследовательский институт диабета, Научный институт Сан-Раффаэле, Милан, Италия.
- 4 Центральный региональный центр детской диабетологии, Департамент здравоохранения, Университетская клиника Маджоре-делла-Карита, Университет Восточного Пьемонта, Новара, Италия.
- 5 CEINGE Advanced Biotechnologies, кафедра молекулярной медицины и медицинской биотехнологии, Неаполитанский университет им. Федерико II, Неаполь, Италия.
- 6 Кафедра педиатрии, Университет медицинских наук, Учебно-исследовательская больница Анкары, Анкара, Турция.
- 7 Кафедра экспериментальной медицины, Университет Тор Вергата, Рим, Италия.

- PMID: 32274916
- DOI: 10.23736/С0026-4946.20.05838-7
Обзор
Dario Iafusco et al. Минерва Педиатр. 2020 авг.
. 2020 авг; 72 (4): 240-249.
doi: 10.23736/S0026-4946.20.05838-7. Epub 2020 9 апр.
Авторы
Дарио Яфуско 1 , Анжела Занфардино 2 , Риккардо Бонфанти 3 , Ивана Раббоне 4 , Надя Тинто 5 , Фернанда Яфуско 5 , Серена Меола 5 , Мария Ф.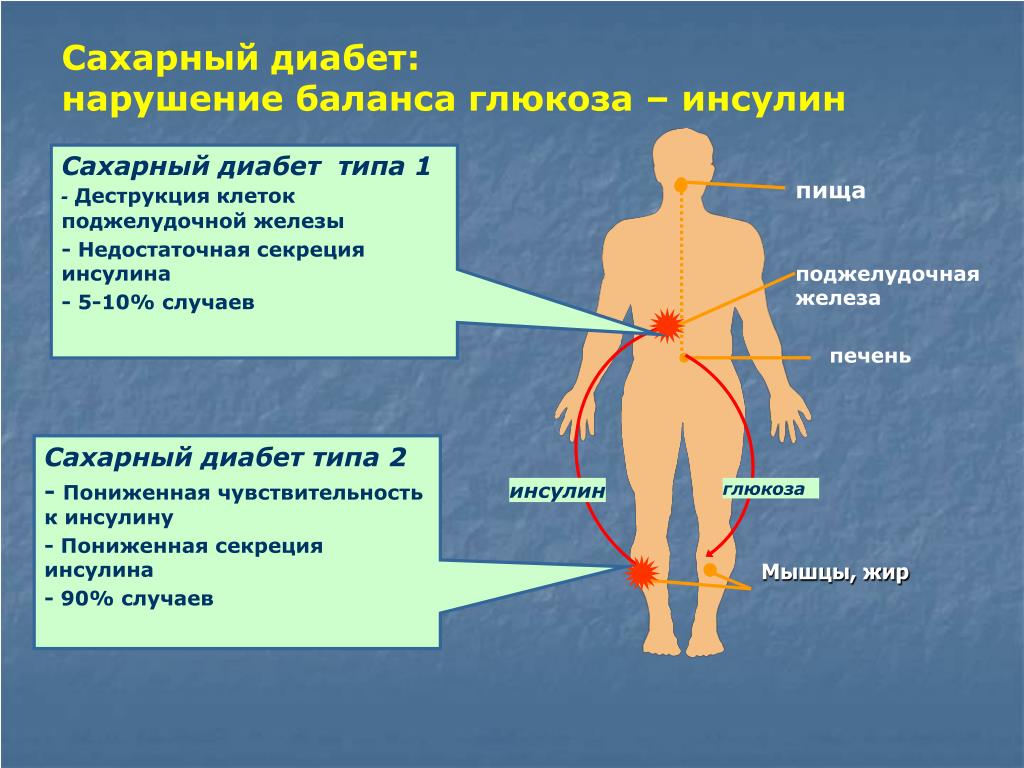 Гиккино 2 , Гульсум Озен 6 , Франческа Касабуро 2 , Алессия Пископо 2 , Эмануэле Миралья-дель-Джудиче 2 , Фабрицио Барбетти 7
Гиккино 2 , Гульсум Озен 6 , Франческа Касабуро 2 , Алессия Пископо 2 , Эмануэле Миралья-дель-Джудиче 2 , Фабрицио Барбетти 7
Принадлежности
- 1 Кафедра педиатрии Университета Кампании «Луиджи Ванвителли», Неаполь, Италия – [email protected].
- 2 Кафедра педиатрии Кампанского университета им. Луиджи Ванвителли, Неаполь, Италия.
- 3 Отделение детской диабетологии, Отделение педиатрии, Научно-исследовательский институт диабета, Научный институт Сан-Раффаэле, Милан, Италия.
- 4 Центральный региональный центр детской диабетологии, Департамент здравоохранения, Университетская клиника Маджоре-делла-Карита, Университет Восточного Пьемонта, Новара, Италия.
- 5 CEINGE Advanced Biotechnologies, кафедра молекулярной медицины и медицинской биотехнологии, Неаполитанский университет им. Федерико II, Неаполь, Италия.
- 6 Кафедра педиатрии, Университет медицинских наук, Учебно-исследовательская больница Анкары, Анкара, Турция.
- 7 Кафедра экспериментальной медицины, Университет Тор Вергата, Рим, Италия.

- PMID: 32274916
- DOI:
10. 23736/С0026-4946.20.05838-7
 23736/С0026-4946.20.05838-7
23736/С0026-4946.20.05838-7Абстрактный
Врожденный сахарный диабет — редкое заболевание, характеризующееся гипергликемией, возникающей вскоре после рождения. Мы определяем «диабет младенческого возраста», если гипергликемия начинается до 6 месяцев жизни. С клинической точки зрения мы различаем два основных типа диабета детского возраста: транзиторный (TNDM), который спонтанно разрешается, и постоянный (PNDM), который требует пожизненного лечения. TNDM может рецидивировать в более позднем возрасте. Около 50% случаев являются преходящими (TNDM) и 50% постоянными. Клинические проявления включают тяжелую задержку внутриутробного развития, гипергликемию и обезвоживание. Сообщается о широком спектре различных сопутствующих клинических признаков, включая лицевой дисморфизм, глухоту и неврологические, сердечные, почечные или мочевыводящие аномалии. Также могут наблюдаться задержка развития и трудности в обучении.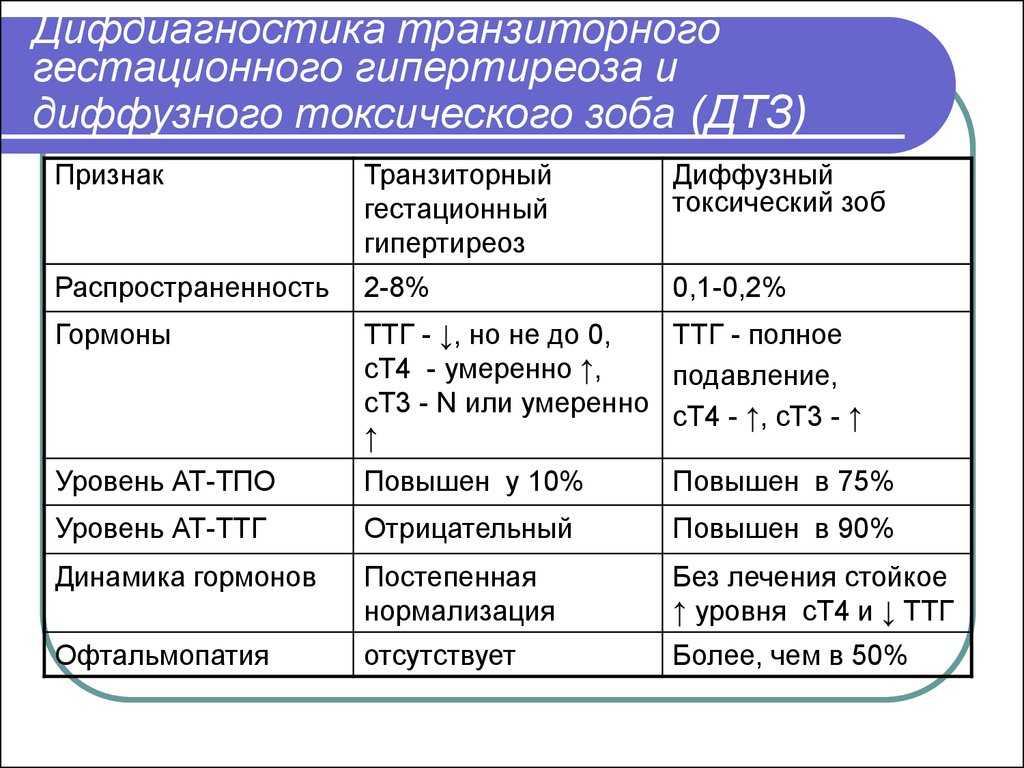 В этой статье мы рассматриваем все причины врожденного диабета и все гены и синдромы, вовлеченные в эту патологию. Открытие патогенеза большинства форм врожденного диабета позволило адаптировать терапию к диагнозу, а при формах альтерации калиевых каналов бета-клеток поджелудочной железы переход с инсулина на глибенкламид per os значительно улучшил качество жизни. Врожденный диабет, хотя и является очень редкой формой, в последние годы был предметом исследований, особенно в области патогенеза и фармакогенетики. Наиболее ярким отличием по сравнению с более частым аутоиммунным диабетом у детей (СД 1 типа) является возможность лечения гипогликемическими средствами и явно меньшая частота хронических осложнений.
В этой статье мы рассматриваем все причины врожденного диабета и все гены и синдромы, вовлеченные в эту патологию. Открытие патогенеза большинства форм врожденного диабета позволило адаптировать терапию к диагнозу, а при формах альтерации калиевых каналов бета-клеток поджелудочной железы переход с инсулина на глибенкламид per os значительно улучшил качество жизни. Врожденный диабет, хотя и является очень редкой формой, в последние годы был предметом исследований, особенно в области патогенеза и фармакогенетики. Наиболее ярким отличием по сравнению с более частым аутоиммунным диабетом у детей (СД 1 типа) является возможность лечения гипогликемическими средствами и явно меньшая частота хронических осложнений.
Похожие статьи
Неонатальный сахарный диабет: заболевание, связанное с несколькими механизмами.
Полак М., Каве Х. Полак М. и др.
Orphanet J Rare Dis. 2007 9 марта; 2:12. дои: 10.1186/1750-1172-2-12.
Orphanet J Rare Dis. 2007.
PMID: 17349054
Бесплатная статья ЧВК.
Обзор.Неонатальный сахарный диабет: хромосомный анализ в транзиторных и постоянных случаях.
Мец С., Каве Х., Бертран А.М., Дефферт С., Геген-Жиру Б., Чернихов П., Полак М.; Французская исследовательская группа NDM. Неонатальный сахарный диабет. Мец С. и соавт. J Педиатр. 2002 г., октябрь; 141 (4): 483-9. doi: 10.1067/mpd.2002.127089. J Педиатр. 2002. PMID: 12378186 Клиническое испытание.
Неонатальный сахарный диабет: обновленная информация о диагностике и лечении.
Lemelman MB, Letourneau L, Greeley SAW. Лемельман М.Б. и соавт. Клин Перинатол.
2018 март;45(1):41-59. doi: 10.1016/j.clp.2017.10.006. Epub 2017 16 декабря.
Клин Перинатол. 2018.
PMID: 29406006
Бесплатная статья ЧВК.
Обзор.Клинические и генетические особенности аргентинских детей с началом диабета в возрасте до 12 месяцев: успешный перевод с инсулина на пероральные препараты сульфонилмочевины.
Табернер П., Фланаган С.Е., Маккей Д.Дж., Эллард С., Таверна М.Дж., Ферраро М. Табернер П. и соавт. Diabetes Res Clin Pract. 2016 июль; 117: 104-10. doi: 10.1016/j.diabres.2016.04.005. Epub 2016 26 апр. Diabetes Res Clin Pract. 2016. PMID: 27329029
Долгосрочное наблюдение транзиторного неонатального сахарного диабета из-за новой гомозиготной мутации c.7734C>T (p.R228C) в гене ZFP57 : рецидив в препубертатном возрасте.
Контбай Т., Атар М., Демирбилек Х. Контбай Т. и др. J Pediatr Endocrinol Metab. 2022 28 февраля; 35 (5): 695-698. doi: 10.1515/jpem-2021-0538. Печать 2022 25 мая. J Pediatr Endocrinol Metab. 2022. PMID: 35218690
 Orphanet J Rare Dis. 2007 9 марта; 2:12. дои: 10.1186/1750-1172-2-12.
Orphanet J Rare Dis. 2007.
PMID: 17349054
Бесплатная статья ЧВК.
Обзор.
Orphanet J Rare Dis. 2007 9 марта; 2:12. дои: 10.1186/1750-1172-2-12.
Orphanet J Rare Dis. 2007.
PMID: 17349054
Бесплатная статья ЧВК.
Обзор. 2018 март;45(1):41-59. doi: 10.1016/j.clp.2017.10.006. Epub 2017 16 декабря.
Клин Перинатол. 2018.
PMID: 29406006
Бесплатная статья ЧВК.
Обзор.
2018 март;45(1):41-59. doi: 10.1016/j.clp.2017.10.006. Epub 2017 16 декабря.
Клин Перинатол. 2018.
PMID: 29406006
Бесплатная статья ЧВК.
Обзор.
Посмотреть все похожие статьи
Типы публикаций
термины MeSH
- 1
- PNDM
- Реестр генетического тестирования: агенезия поджелудочной железы 1
- Реестр генетического тестирования: постоянный неонатальный сахарный диабет
- Постоянный сахарный диабет новорожденных
- Информационный поиск по болезням
- Национальная организация редких заболеваний (NORD)
- ClinicalTrials.gov
- САХАРНЫЙ ДИАБЕТ ПОСТОЯННЫЙ НОВОРОЖДЕННЫЙ 1
- Агенезия поджелудочной железы 1
- PubMed
- Барбарини Д.С., Хаслингер В., Шмидт К., Патч А.М., Мюллер Г., Симма Б. Неонатальный сахарный диабет вследствие агенезии поджелудочной железы: новый клинический случай и обзор литература. Педиатр Диабет. 2009 ноябрь;10(7):487-91. дои: 10.1111/j.1399-5448.2009.00523.х. Epub 2009, 3 июня. Цитирование в PubMed .
- Эдгхилл Э.Л., Фланаган С.Э., Эллард С. Постоянный неонатальный диабет из-за активирующие мутации в ABCC8 и KCNJ11. Rev Endocr Metab Disord. 2010 Сен;11(3):193-8. doi: 10.1007/s11154-010-9149-x. Цитата в PubMed
- Edghill EL, Flanagan SE, Patch AM, Boustred C, Parrish A, Shields B, Shepherd
М.Х., Хуссейн К. , Капур Р.Р., Малеки М., Макдональд М.Дж., Стой Дж., Штайнер Д.Ф., Филипсон
Л.Х., Белл Г.И.; Международная совместная группа по неонатальному диабету; Хаттерсли АТ,
Эллард С. Скрининг мутаций инсулина у 1044 пациентов с диабетом: мутации
в гене INS являются частой причиной неонатального диабета, но редкой причиной
сахарный диабет, диагностированный в детстве или во взрослом возрасте. Диабет. 2008 г., апрель 57(4):1034-42.
дои: 10.2337/db07-1405. Epub 2007, 27 декабря. Цитирование на PubMed
- Эллард С., Фланаган С.Э., Жирар К.А., Патч AM, Харрис Л.В., Пэрриш А., Эдгхилл Э.Л., Mackay DJ, Proks P, Shimomura K, Haberland H, Carson DJ, Shield JP, Hattersley AT, Эшкрофт FM. Постоянный неонатальный диабет, вызванный доминантным, рецессивным или сложные гетерозиготные мутации SUR1 с противоположными функциональными эффектами. Ам Джей Хам Жене. 2007 г., август; 81 (2): 375-82. дои: 10.1086/519174. Epub 2007, 29 июня. Цитирование в PubMed или бесплатная статья на PubMed Central .
- Фланаган С.Э., Клоин С., Белланн-Шантело С., де Лонле П., Харрис Л.В., Глойн AL, Эллард С. Обновление мутаций в генах, кодирующих бета-клетки поджелудочной железы. Субъединицы канала K (АТФ) Kir6.2 (KCNJ11) и рецептор 1 сульфонилмочевины (ABCC8) в сахарный диабет и гиперинсулинизм. Хум Мутат. 2009 г.30 февраля (2): 170-80. дои: 10.1002/гуму.20838. Цитата в PubMed
- Фланаган С.Е., Эдгхилл Э.Л., Глойн А.Л., Эллард С., Хаттерсли А.Т. Мутации в KCNJ11, который кодирует Kir6.2, является частой причиной диабета, диагностируемого в первые 6 мес жизни, при этом фенотип определяется генотипом. Диабетология. 2006 г., июнь; 49 (6): 1190-7. doi: 10.1007/s00125-006-0246-z. Epub 2006, 12 апреля. Цитирование в PubMed .
- Malecki MT, Mlynarski W. Моногенный диабет: последствия для терапии редких виды болезни. Сахарный диабет Ожирение Metab. 2008 авг; 10 (8): 607-16. дои: 10.1111/j.1463-1326.2007.00736.х. Epub 2007 6 мая. Цитирование в PubMed
- Осбак К. К., Колклаф К., Сен-Мартен К., Бир Н.Л., Белланн-Шантелот К., Эллард
С, Глойн А.Л. Обновленная информация о мутациях глюкокиназы (GCK), которые вызывают наступление зрелости
сахарный диабет молодых, перманентный неонатальный диабет и гиперинсулинемический
гипогликемия. Хум Мутат. 2009 ноябрь;30(11):1512-26. doi: 10.1002/humu.21110. Цитата в PubMed
- Полак М., Кейв Х. Неонатальный сахарный диабет: заболевание, связанное с множественными механизмы. Orphanet J Rare Dis. 2007 9 марта;2:12. дои: 10.1186/1750-1172-2-12. Цитирование в PubMed или бесплатная статья в PubMed Central
- Рубио-Кабесас О., Клупа Т., Малецки М.Т.; Консорциум CEED3. Постоянный неонатальный сахарный диабет — важность дифференциальной диагностики сахарного диабета у новорожденных и младенцев. Евро Джей Клин Инвест. 2011 март; 41(3):323-33. дои: 10.1111/j.1365-2362.2010.02409.х. Epub 2010, 4 ноября. Цитирование в PubMed .
- Стой Дж., Штайнер Д.
вещества
Перманентный неонатальный сахарный диабет: MedlinePlus Genetics
Описание
Перманентный неонатальный сахарный диабет — это тип диабета, который впервые появляется в течение первых 6 месяцев жизни и сохраняется на протяжении всей жизни. Эта форма диабета характеризуется высоким уровнем сахара в крови (гипергликемия) в результате дефицита гормона инсулина. Инсулин контролирует, сколько глюкозы (разновидности сахара) передается из крови в клетки для преобразования в энергию.
Инсулин контролирует, сколько глюкозы (разновидности сахара) передается из крови в клетки для преобразования в энергию.
Лица с перманентным неонатальным сахарным диабетом имеют медленный рост до рождения (задержка внутриутробного развития). У пострадавших младенцев наблюдается гипергликемия и чрезмерная потеря жидкости (обезвоживание), они не могут набирать вес и расти с ожидаемой скоростью (отставание в развитии).
В некоторых случаях люди с перманентным неонатальным сахарным диабетом также имеют определенные неврологические проблемы, включая задержку развития и повторяющиеся припадки (эпилепсия). Эта комбинация задержки развития, эпилепсии и неонатального диабета называется синдромом DEND. Промежуточный синдром DEND представляет собой аналогичную комбинацию, но с более легкой задержкой развития и без эпилепсии.
У небольшого числа лиц с перманентным неонатальным сахарным диабетом поджелудочная железа недоразвита. Поскольку поджелудочная железа вырабатывает пищеварительные ферменты, а также секретирует инсулин и другие гормоны, у больных возникают проблемы с пищеварением, такие как жирный стул и неспособность усваивать жирорастворимые витамины.
Частота
Приблизительно у 1 из 400 000 младенцев диагностируется сахарный диабет в первые несколько месяцев жизни. Однако примерно у половины этих детей это состояние носит преходящий характер и проходит само по себе к 18 месяцам. Остальные считаются больными перманентным неонатальным сахарным диабетом.
Причины
Постоянный сахарный диабет новорожденных может быть вызван мутациями в нескольких генах.
Около 30 процентов лиц с перманентным неонатальным сахарным диабетом имеют мутации в KCNJ11 ген. Еще 20 процентов людей с перманентным неонатальным сахарным диабетом имеют мутации в гене ABCC8 . Эти гены предоставляют инструкции для создания частей (субъединиц) АТФ-чувствительного калиевого (К-АТФ) канала. Каждый канал K-ATP состоит из восьми субъединиц, четыре из которых продуцируются геном KCNJ11 и четыре из гена ABCC8 .
Каналы K-АТФ обнаружены через клеточные мембраны в секретирующих инсулин бета-клетках поджелудочной железы. Эти каналы открываются и закрываются в зависимости от количества глюкозы в кровотоке. Закрытие каналов в ответ на повышение уровня глюкозы вызывает высвобождение инсулина из бета-клеток в кровоток, что помогает контролировать уровень сахара в крови.
Эти каналы открываются и закрываются в зависимости от количества глюкозы в кровотоке. Закрытие каналов в ответ на повышение уровня глюкозы вызывает высвобождение инсулина из бета-клеток в кровоток, что помогает контролировать уровень сахара в крови.
Мутации в гене KCNJ11 или ABCC8 , которые вызывают необратимый неонатальный сахарный диабет, приводят к тому, что каналы K-АТФ не закрываются, что приводит к снижению секреции инсулина бета-клетками и нарушению контроля уровня сахара в крови.
Мутации в гене INS , отвечающем за выработку инсулина, были выявлены примерно у 20 процентов лиц с перманентным неонатальным сахарным диабетом. Инсулин производится в форме предшественника, называемого проинсулином, который состоит из одной цепи белковых строительных блоков (аминокислот). Цепь проинсулина разрезается (расщепляется) с образованием отдельных частей, называемых цепями А и В, которые соединяются вместе соединениями, называемыми дисульфидными связями, с образованием инсулина.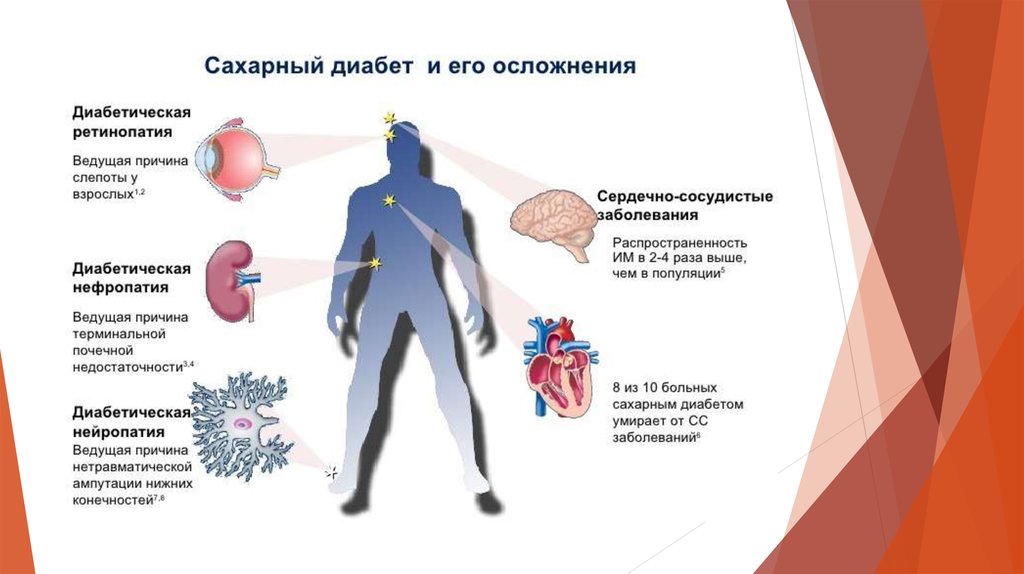 Мутации в 9Считается, что ген 0249 INS нарушает расщепление цепи проинсулина или связывание цепей А и В с образованием инсулина, что приводит к нарушению контроля уровня сахара в крови.
Мутации в 9Считается, что ген 0249 INS нарушает расщепление цепи проинсулина или связывание цепей А и В с образованием инсулина, что приводит к нарушению контроля уровня сахара в крови.
Постоянный сахарный диабет новорожденных также может быть вызван мутациями в других генах, некоторые из которых не были идентифицированы.
Наследование
Перманентный неонатальный сахарный диабет может иметь различные типы наследования.
Когда это состояние вызвано мутациями в гене Ген KCNJ11 или INS наследуется по аутосомно-доминантному типу, что означает, что одной копии измененного гена в каждой клетке достаточно, чтобы вызвать заболевание. Примерно в 90 процентах случаев это состояние возникает в результате новых мутаций в гене и возникает у людей, в семье которых не было этого заболевания. В остальных случаях больной наследует мутацию от одного больного родителя.
Когда постоянный сахарный диабет новорожденных вызван мутациями в ABCC8 , он может наследоваться либо по аутосомно-доминантному, либо по аутосомно-рецессивному типу. При аутосомно-рецессивном наследовании обе копии гена в каждой клетке имеют мутации. Каждый из родителей человека с аутосомно-рецессивным заболеванием несет по одной копии мутировавшего гена, но обычно у них нет признаков и симптомов заболевания.
При аутосомно-рецессивном наследовании обе копии гена в каждой клетке имеют мутации. Каждый из родителей человека с аутосомно-рецессивным заболеванием несет по одной копии мутировавшего гена, но обычно у них нет признаков и симптомов заболевания.
Реже заболевание вызывается мутациями в других генах, и в этих случаях оно также наследуется по аутосомно-рецессивному типу.
Другие названия для этого состояния
Дополнительная информация и ресурсы
Информация о генетическом тестировании
Информационный центр генетических и редких заболеваний
Ресурсы поддержки пациентов и защиты интересов
Научные исследования от ClinicalTrials.
 gov
govКаталог генов и болезней от OMIM
Научные статьи в PubMed
Ссылки
 , Капур Р.Р., Малеки М., Макдональд М.Дж., Стой Дж., Штайнер Д.Ф., Филипсон
Л.Х., Белл Г.И.; Международная совместная группа по неонатальному диабету; Хаттерсли АТ,
Эллард С. Скрининг мутаций инсулина у 1044 пациентов с диабетом: мутации
в гене INS являются частой причиной неонатального диабета, но редкой причиной
сахарный диабет, диагностированный в детстве или во взрослом возрасте. Диабет. 2008 г., апрель 57(4):1034-42.
дои: 10.2337/db07-1405. Epub 2007, 27 декабря. Цитирование на PubMed
, Капур Р.Р., Малеки М., Макдональд М.Дж., Стой Дж., Штайнер Д.Ф., Филипсон
Л.Х., Белл Г.И.; Международная совместная группа по неонатальному диабету; Хаттерсли АТ,
Эллард С. Скрининг мутаций инсулина у 1044 пациентов с диабетом: мутации
в гене INS являются частой причиной неонатального диабета, но редкой причиной
сахарный диабет, диагностированный в детстве или во взрослом возрасте. Диабет. 2008 г., апрель 57(4):1034-42.
дои: 10.2337/db07-1405. Epub 2007, 27 декабря. Цитирование на PubMed
 К., Колклаф К., Сен-Мартен К., Бир Н.Л., Белланн-Шантелот К., Эллард
С, Глойн А.Л. Обновленная информация о мутациях глюкокиназы (GCK), которые вызывают наступление зрелости
сахарный диабет молодых, перманентный неонатальный диабет и гиперинсулинемический
гипогликемия. Хум Мутат. 2009 ноябрь;30(11):1512-26. doi: 10.1002/humu.21110. Цитата в PubMed
К., Колклаф К., Сен-Мартен К., Бир Н.Л., Белланн-Шантелот К., Эллард
С, Глойн А.Л. Обновленная информация о мутациях глюкокиназы (GCK), которые вызывают наступление зрелости
сахарный диабет молодых, перманентный неонатальный диабет и гиперинсулинемический
гипогликемия. Хум Мутат. 2009 ноябрь;30(11):1512-26. doi: 10.1002/humu.21110. Цитата в PubMed