
Миопатия миотубулярная – Врожденная миопатия > Архив — Клинические протоколы МЗ РК
Врожденная миопатия > Архив — Клинические протоколы МЗ РК
Диагностические критерии
Жалобы и анамнез: заболевание проявляется с рождения или в раннем детском возрасте; задержка в моторном развитии, гипотрофия, мышечная слабость, снижение тонуса и мышечной силы, нарушение походки.
Физикальные обследования
Болезнь центрального стержня
Наследуется по аутосомно-доминантному типу, характеризуется специфическими морфологическими изменениями, при которых в мышечных волокнах в центральной части имеются участки, заполненные компактными миофибриллами — «стержни». Диаметр волокон сохраняется, иногда он даже несколько увеличен.
Клиническая картина характеризуется более или менее значительной мышечной слабостью, преобладающей в проксимальных отделах конечностей, особенно нижних, и диффузной мышечной гипотонией.
Типично отставание в двигательном развитии при нормальном интеллекте. Мышечные атрофии выражены умеренно, сухожильные рефлексы сохранены или нерезко снижены.
Болезнь центрального ядра (миотубулярная миопатия)
Типы наследования: аутосомно-доминантный, аутосомно-рециссивный, рециссивный, сцепленный с Х-хромосомой. Эти обстоятельства говорят о генетической гетерогенности миотубулярной миопатии. Морфологические изменения при миотубулярной миопатии характеризуются уменьшением диаметра мышечных волокон. В центре этих малых волокон миофибриллы отсутствуют или очень рыхло упакованы.
Гистохимические реакции выявляют в центральных областях высокую активность окислительных ферментов. Измененные мышечные волокна в центре очень напоминают эмбриональные мышечные клетки или миотубулы. В связи с этим заболевание получило название митубулярной миопатии. Другое ее название — центронуклеарная, что обусловлено скоплением в центральной части мышечного волокна группы ядер.
Течение заболевания часто стационарное, но может иметь место регресс симптомов или со слабым прогрессированием. В большинстве случаев клинические симптомы отмечаются с первых дней жизни, но иногда проявляются впервые у взрослых.
Миопатия с врожденной диспропорцией типов волокон
Мышечные волокна делятся на два типа: I тип — красные волокна, II тип — белые. В последнее время II тип подразделяют на II A и II Б типы. В норме типы I и II имеют одинаковые размеры, их соотношение неодинаково в различных мышечных группах, но это различие довольно постоянно. При изучении врожденных форм миопатий с клинической картиной «floppy baby», было показано, что у некоторых больных патоморфологическое исследование выявляет единственный дефект — уменьшение в размерах типа I мышечных волокон и их преобладание, и сохранность величины увеличения в размере типа II волокон.
Термин «врожденная миопатия с диспропорцией типов волокон» используется для описания новорожденных с гипотонией и иногда с артрогрипозом.
Немалиновая (нитеобразная) миопатия — наиболее частая форма. Патология состоит в наличии нитеобразных структур вдоль всего длинника пораженных мышечных клеток. Заболевание передается как по аутосомно-рециссивному, так и по аутосомно-доминантному типу. В основном семейные случаи заболевания, но все чаще спорадические случаи.
Тяжесть состояния широко варьирует — от очень легкой до выраженной слабости с гипотрофией, приводящей к инвалидности. Больные отстают в двигательном развитии, позже начинают сидеть, ходить. Характерно сочетание мышечных симптомов с диспластическими изменениями скелета в виде вытянутой формы лицевого черепа, ретрогнатии «готического неба», деформации грудной клетки, позвоночника.
При биохимическом исследовании находят некоторое повышение активности креатинфосфокиназы в сыворотке крови.
Митохондриальные миопатии — группа миопатий, в которых специфическим и единственным морфологическим нарушением является патология митохондрий. Наследуются по аутосомно-рециссивному типу. Эта группа миопатий является гетерогенной, поскольку она представлена различными типами нарушения митохондрий.
Плеокониальная миопатия — характеризуется эпизодическими приступами, начинающимися с жажды, рвоты, выраженной потребности в соли, профузного пота. Развивается мышечная слабость вплоть до вялой тетраплегии с отсутствием сухожильных рефлексов. Нарушается также речь и глотание. Приступ длится 10-14 дней. Выраженная мышечная слабость наблюдается с рождения, с возрастом может даже несколько уменьшаться. При морфологическом исследовании в мышечных волокнах обнаружены митохондрии гигантских размеров.
Илеокониальная миопатия
Врожденная мышечная дистрофия — гипотония и артрогрипоз определяются с рождения. Сухожильные рефлексы либо сохранны, либо отсутствуют, но из-за контрактур суставов их бывает трудно оценить. Врожденные контрактуры могут вовлекать различные суставы, но наиболее часто они приводят к кривошее и косолапости. Характерный симптом — врожденный вывих бедра.
Инструментальные исследования:
1. Компьютерная томография головного мозга для исключения органического поражения.
2. Электроэнцефалография (ЭЭГ) — изменения ЭЭГ неспецифичны.
3. Электромиография (ЭМГ) — выявляет первичный мышечный дефект, снижение амплитуды ЭМГ максимального напряжения, длительность потенциалов ДЕ уменьшена, увеличение числа полифазных потенциалов без увеличения их длительности, иногда ЭМГ имеет смешанный тип.
4. Магниторезонансная томография (МРТ) головного мозга — в настоящее время является методом выбора при диагностике пороков и аномалий и дегенеративных изменений головного мозга.
5. Нейросонография — выявляет наличие структурных изменений, расширение желудочковой системы, пороки и дизгенезии до закрытия родничка.
6. ЭКГ — изменения, указывающие на обменно-дистрофические изменения в миокарде.
Показания для консультаций специалистов:
1. Логопед — для выявления нарушения речи, дизартрий.
2. Психолог — для определения психического статуса ребенка.
3. Ортопед — для выявления контрактур, костных деформаций.
4. Протезист — для подбора ортопедической обуви, фиксирующих лонгет.
5. Окулист — осмотр глазного дна, выявление страбизма, нарушений зрения.
6. Кардиолог — выявление обменно-дистрофических и диспластических изменений сердца.
7. ЛОР-сурдолог — для определения тугоухости.
Минимум обследования при направлении в стационар:
1. Общий анализ крови.
2. Общий анализ мочи.
3. Кал на яйца глист.
4. АЛТ.
5. АСТ.
Основные диагностические мероприятия:
1. Общий анализ крови.
2. Общий анализ мочи.
3. Логопед.
4. Психолог.
5. Окулист.
6. Ортопед.
7. ЭЭГ.
8. Врач ЛФК.
9. Физиотерапевт.
10. Электромиография (ЭМГ).
11. ЭКГ.
12. Кардиолог.
13. ЛОР-сурдолог.
14. Определение креатинкиназы в сыворотке крови.
Перечень дополнительных диагностических мероприятий:
1. Компьютерная томография головного мозга.
2. Нейросонография.
3. Нейрохирург.
4. Протезист.
5. МРТ головного мозга.
6. ЭхоКГ.
7. R-графия грудной клетки.
8. УЗИ органов брюшной полости.
9. Генетик.
10. ЛОР сурдолог.
11. ЭЭГ.
12. ИФА на ТТГ.
13. Эндокринолог.
14. Мышечная биопсия.
diseases.medelement.comМиотубулярная миопатия (частный случай) — Мед Др
Нами наблюдалась семья с миотубулярной миопатией, имеющей ряд особенностей. Одной из них является четкое наследование по рецессивному, сцепленному с Х-хромосомой типу.
Родословная семьи больного с миотубулярной миопатией
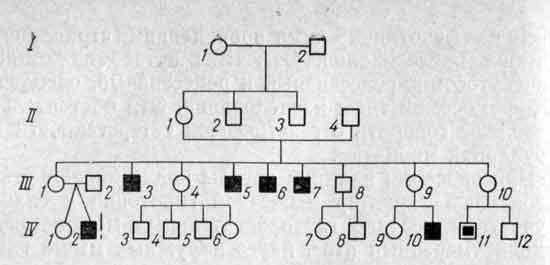
Х-Сцепленный тип наследования. Обозначения те же, что и на рисунке
Родословная семьи Д-вых.
Пробанд (IV-II) — 13 летний мальчик — родился от 1-й беременности. Шевеление плода было слабое. Родился в срок, но в состоянии асфиксии. С первых дней жизни наблюдались вялость, резкое ограничение объема движений в конечностях. Голос был очень слабым, дыхание поверхностным, самостоятельное глотание было невозможным, в связи с чем в первые 5 мес ребенка кормили через зонд.
Становление двигательных функций было замедленным — г голову стал держать с 7 мес, сидеть с 9 мес, ходить с 1,5 лет, походка была валкой, часто падал, с трудом поднимался с пола. До 10 лет постепенно наблюдалась компенсация и улучшение активных движений и мышечной силы, однако в последние 3 года несколько увеличилась слабость.
Родители и 11-летний брат здоровы.
Мальчик пониженного питания. Отмечается нерезко выраженная деформация грудины. Интеллект сохранен. Имеется резкое ограничение движений глазных яблок вверх и в стороны. Птоза нет. Умеренная слабость мимической мускулатуры. Диффузная гипотрофия мышц шеи, туловища, тазового и плечевого поясов, конечностей.
Генерализованная мышечная слабость, особенно в сгибателях шеи, мышцах плечевого и тазового поясов. При вставании с пола пользуется миопатическими приемами. Нерезко выражены сгибательные контрактуры в локтевых суставах. Сухожильные рефлексы не вызываются, за исключением ахилловых, которые средней живости. Клинический анализ крови и мочи без патологии.
Отмечается четкая гиперферментемия: альдолаза 26 ЕД (норма до 6 ЕД), ЛДГ-550 МЕ/мл (норма — до 180 МЕ/ /мл), КФК — 650 ME (норма — до 100 ME), ACT — 108 ЕД, AЛT — 91 ЕД. Креатин в суточной моче — 0,38 ммоль/сут, креатинин — 6,6 ммоль/ сут.
ЭМГ: при исследовании игольчатыми электродами в покое спонтанной активности не выявлено. При максимальном усилии зарегистрирована высокоамплитудная интерференционная кривая. Двигательные единицы удлинены, отмечается большое число полифазных потенциалов. Данные ЭМГ позволяют предполагать неврогенный уровень страдания.
При исследовании биоптата четырехглавой мышцы бедра отмечена резко выраженная разнокалиберность мышечных волокон (5 — 70 ммк). В 35% волокон имеется центральное расположение ядер (1 — 5), которые имеют тот же вид, что и субсарколеммные ядра, или они светлые, пузырчатые, содержат четко видимое ядрышко. Количество субсарколеммных ядер в этих волокнах уменьшено или они отсутствуют.
При обработке на окислительно-восстановительные ферменты (сукцинат- , малат- , лактатдегидрогеназу) общая ферментативная активность, высокая, однако четкой дифференцировки на типы волокон не отмечено. В пораженных волокнах видны центральные или парацентральные участки, в которых снижена активность окислительно-восстановительных ферментов. При обработке на АТФ-азу выявлены зоны, лишенные этой активности.
Больной IV-10, 10 лет, родился от 2-й беременности в срок, но в состоянии асфиксии. Проводились интенсивные реанимационные мероприятия. На 114-день у ребенка развилась пневмония, в связи с неоднократными рецидивами которой в течение 9 мес он непрерывно находился в стационаре, получая интенсивную антибиотикотерапию.
С первых дней жизни отмечалась выраженная вялость движений. Сидеть стал с 10 мес, ходить — с 1,5 лет. Походка была валкой, часто падал. В последующие годы отмечена постепенная компенсация двигательного дефекта. В 8-месячном возрасте наблюдался единичный судорожный эпилептиформный припадок.
Родители и 7-летняя сестра мальчика здоровы.
Двоюродные сибсы с миотубулярной миопатией
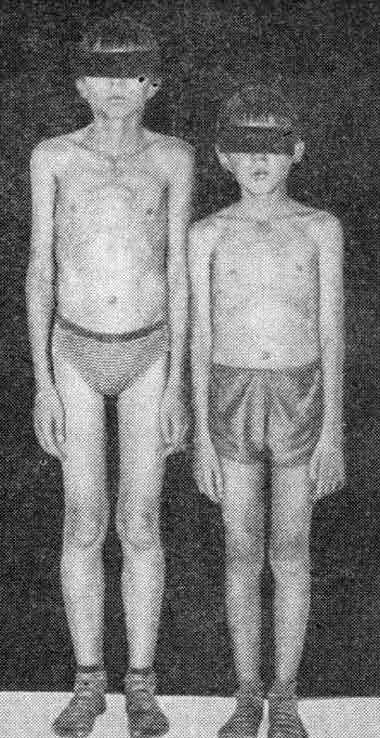
Диффузная мышечная гипотрофия.
Объективно больной пониженного питания. Умеренная деформация грудины. Гипертелоризм. Птоза нет. Резкое ограничение движений глазных яблок вверх, недоведение глаз в стороны, диплопии нет, движение вниз в полном объеме. Ослаблена мимическая мускулатура. Речь с носовым оттенком. Глотание не нарушено. Отмечаются умеренная гипотрофия и слабость мышц туловища, плечевого и тазового поясов, конечностей.
Выраженная слабость в сгибателях шеи: находясь в положении лежа на спине, не может согнуть голову вперед. Сухожильные рефлексы отсутствуют, кроме ахилловых, которые вызываются, но снижены.
КФК сыворотки крови — 1300 ME, ЛДГ — 550 ЕД, альдолаза — 35 ЕД, ACT — 134 ЕД, АЛТ — 130 ЕД. Креатина в суточной моче 3,04 ммоль, креатинина — 2,9 ммоль.
ЭМГ при использовании игольчатых электродов выявила спонтанную активность в виде фибрилляций частотой до 15 Гц и амплитудой 50 — 100 мкВ. При максимальном усилии регистрируется интерференционная высокоамплитудная электрическая активность (до 2000 — 3000 мкВ). Длительность ПД ДЕ в среднем не изменена, однако имеется большое количество ПД ДЕ как длинных (> 113 мс), так и коротких (< 5 мс) с амплитудой 2000 — 2500 мкВ. Отмечено 35% полифазных потенциалов.
Заключение: результаты ЭМГ-ого исследования указывают на спинальный уровень поражения.
Исследование биоптатов дельтовидной мышцы выявило изменения, аналогичные таковым у больного IV-11.
Больной IV-2, 22 лет, родился в срок в разнояйцовой двойне в состоянии асфиксии. С первых дней жизни отмечалась общая вялость, наблюдалось резкое отставание в становлении двигательных функций.
Однако постепенно с годами двигательный дефект уменьшался, к 18 годам чувствовал себя практически здоровым, был призван в ряды армии. Во время службы выполнял обязанности водителя автомашины. С 8 лет страдал приступами «уже виденного», по поводу чего принимал финлепсин. Больших судорожных приступов не отмечалось.
Объективно: двусторонний полуптоз без ограничения движений глазных яблок. Умеренная слабость мимической мускулатуры. Четкое снижение силы в сгибателях шеи. В остальных группах мышц сила достаточная. Сухожильные рефлексы живые. Активность сывороточной КФК — 48 ЕД.
Больные III-3, III-5, III-6, III-7 — все 4 мальчика — родились в срок в асфиксии. У всех отмечались выраженная слабость и поверхностное дыхание. Во всех случаях наступил летальный исход в первые 1 — 3 нед жизни в результате пневмонии.
Таким образом, в этой семье в двух поколениях имелось 7 больных со стереотипной картиной врожденной резко выраженной вялости и мышечной слабости с поражением дыхательной мускулатуры (клиника «вялого ребенка»).
Несмотря на тяжесть поражения в ранние сроки жизни, если удавалось спасти больных от пневмонии, то в последующие годы наступала постепенная компенсация двигательного дефекта. У больного 22 лет имеется уже минимальная недостаточность, он полностью адаптирован и выполняет достаточно тяжелую физическую работу. У всех больных имело место поражение глазодвигательных и мимических мышц, а также сгибателей шеи.
Обращает внимание достаточно высокая гиперферментемия у младших больных, низкая экскреция креатинина, наличие креатина в моче, что характерно для первичного поражения мышц. Вместе с тем ЭМГ-ское исследование показало заинтересованность спинального уровня. Подобные изменения отмечены и другими авторами [Wijugaarden G. К. et al., 1969; Radu Н. et al., 1977].
Данные морфологического исследования четко указывают на типичную миотубулярную миопатию. Постепенная компенсация дефекта с возрастом может быть расценена как позднее постепенное созревание мышечной ткани.
«Нервно-мышечные болезни»,
Б.М.Гехт, Н.А.Ильина
Читайте далее:
www.meddr.ru
Врожденная миопатия — причины, симптомы, диагностика и лечение
Врожденная миопатия — врожденное заболевание, обусловленное генетически детерминированными нарушениями в строении мышечной ткани. Врожденная миопатия проявляется диффузной мышечной слабостью и снижением мышечного тонуса, выраженность которых значительно варьирует в зависимости от вида миопатии. В тяжелых случаях врожденная миопатия может привести к гибели ребенка от дыхательной недостаточности. Диагностируется врожденная миопатия в основном по результатам морфологического исследования образцов, полученных при биопсии мышц; электромиография, эргометрия и исследование мышечного тонуса имеют лишь вспомогательное значение. Врожденная миопатия может потребовать мероприятий по борьбе с дыхательными нарушениями, обеспечению зондового питания, коррекции имеющихся ортопедических деформаций и пр.
Общие сведения
Термин «врожденная миопатия» применяется в неврологии в отношении целой группы достаточно редких наследственных болезней, которые имеют сходную клиническую картину и дифференцируются лишь по специфическим морфологическим изменениям в строении мышечной ткани. К наиболее часто встречаемым видам врожденной миопатии относятся: болезнь центрального стержня, немалиновая миопатия, миотубулярная миопатия, миопатия с множественными стержнями и врожденная диспропорциональность типов мышечных волокон.
Врожденная миопатия является генетически обусловленным заболеванием. В зависимости от вида миопатии аномалия может локализоваться в различных локусах хромосом и передаваться по наследству доминантно, рецессивно или сцеплено с Х-хромосомой. Наличие аномального гена приводит к нарушению синтеза того или иного белка, входящего в структуру мышечной ткани. В результате изменяется строение мышечных волокон, что негативно отражается на их сократительной способности и приводит к генерализованной мышечной слабости. Обычно врожденная миопатия проявляется в раннем детском возрасте. Ее симптомы сохраняются в течение всей жизни пациента. В большинстве случаев врожденная миопатия характеризуется доброкачественным течением со слабым прогрессированием или вовсе без него.
Врожденная миопатия
Классификация врожденной миопатии
В основу классификации врожденной миопатии были положены 2 признака: наличие информации о локализации генной аномалии и данных о том, какой именно белок мышечной ткани является дефектным. В соответствии с этим выделяется врожденная миопатия с известным мутантным геном и определенным дефектным белком (немалиновая миопатия, болезнь центрального стержня, миотубулярная миопатия), врожденная миопатия с неопределенным дефектным белком, но установленным мутантным геном (десмин-связанная и актинзависимая миопатии) и врожденная миопатия, для которой неизвестными остаются и ген, и дефектный белок (врожденная диспропорция типов волокон, центронуклеарная миопатия, миопатия с множественными стержнями).
Симптомы врожденной миопатии
Врожденная миопатия в первые месяцы жизни ребенка характеризуется наличием синдрома «вялого ребенка»: диффузным снижением мышечного тонуса, легкой мышечной слабостью, плохим развитием мускулатуры и ослабленным сосанием. По мере развития ребенка мышечная слабость становится более заметной. Она проявляется невозможностью подняться с пола, залезть на стул, затруднениями при ходьбе и других действиях, которые без проблем выполняют другие дети того же возраста. Мышечная слабость при врожденной миопатии может быть выражена в различной степени. Обычно не наблюдается ее существенное прогрессирование. В тяжелых случаях ребенок так и не может встать на ноги и вынужден всю жизнь передвигаться на каталке. Но те навыки, которые были им приобретены, уже не утрачиваются.
Наибольшая опасность врожденной миопатии связана со слабостью дыхательной мускулатуры. При умеренной мышечной слабости отмечается постепенное развитие дыхательной недостаточности, частые бронхо-легочные заболевания (бронхит, очаговая пневмония, застойная пневмония и др.). Выраженная слабость дыхательных мышц может приводить к быстрому развитию дыхательной недостаточности и гибели ребенка в младенческом возрасте.
В некоторых случаях врожденная миопатия сочетается с дисморфичными чертами (высокое небо, удлиненная и узкая форма лица) и скелетными аномалиями (кифоз, сколиоз, косолапость, грудь сапожника, врожденный вывих бедра).
Характеристика отдельных видов врожденной миопатии
Болезнь центрального стержня наследуется аутосомно-доминантно, известны также отдельные спорадические случаи заболевания. Врожденная миопатия этого вида проявляется задержкой двигательного развития в период первого года жизни, реже обнаруживается у взрослых пациентов, часто сопровождается слабостью мимической мускулатуры. Характерен небольшой рост больных и хрупкая фигура, наличие скелетных деформаций. У пациентов с этим видом врожденной миопатии отмечается повышенный риск возникновения злокачественной гипертермии. В биоптате мышечной ткани выявляют мышечные волокна с единичными или множественными зонами асептического некроза.
Немалиновая врожденная миопатия включает актинопатию, небулинопатию, тропомиозинопатию и тропонинопатию. Ее наследование происходит чаще по аутосомно-доминантному принципу, но также встречается рецессивное наследование и спорадические случаи заболеваемости. Классическая форма немалиновой врожденной миопатии характеризуется синдромом вялого ребенка. Тяжелая форма проявляется еще во внутриутробном периоде в виде акинезии плода, а при рождении ребенка — тяжелыми двигательными нарушениями, слабостью мышц лица и дыхательной недостаточностью. Легкая форма этого типа врожденной миопатии диагностируется после периода раннего детства, иногда — в подростковом возрасте, и протекает без слабости лицевой мускулатуры. Существует также специфическая форма немалиновой врожденной миопатии, при которой возможно развитие офтальмоплегии, кардиомиопатии, синдрома ригидного позвоночника. Морфологическое исследование обнаруживает наличие в мышцах характерных палочко- или нитеподобных телец.
Миотубулярная врожденная миопатия чаще наследуется как аутосомная, при которой мышечная слабость выражена в легкой степени и может наблюдаться как у девочек, так и у мальчиков. Х-сцепленная миотубулярная врожденная миопатия поражает только лиц мужского пола и характеризуется более тяжелым течением со слабостью лицевых мышц, расстройством глотания и дыхательной функции. В биоптате мышечной ткани преобладает поражение волокон I типа. Отмечается центральное расположение ядер миоцитов, что соответствует мышечной ткани эмбриона на 8-10 недели беременности. В связи с эти большинство исследователей рассматривают миотубулярную миопатию как результат недоразвития мышечной ткани.
Миопатия с множественными стержнями чаще наблюдается как аутосомно-рецессивное заболевание, хотя возможен и доминантный тип наследования. Типична мышечная слабость в проксимальных отделах, которая наблюдается в грудном возрасте. Намного реже заболевание дебютирует в более старшем возрасте. В таких случаях отмечается генерализованная мышечная слабость. В мышечном биоптате определяются клетки с отсутствием митохондрий, деструкция сакромеров и гипотрофия мышечных волокон.
Врожденная диспропорция типов мышечных волокон проявляется генерализованной слабостью мышц, в том числе и лицевых, мышечной гипотонией, аномалиями скелета. Тип наследования этой врожденной миопатии пока не установлен. В биоптате мышц наблюдается увеличение количества и малый размер волокон I типа на фоне гипертрофии или нормального размера волокон II типа.
Диагностика врожденной миопатии
В легких случаях только прицельный осмотр с исследованием мышечной силы и тонуса позволяет неврологу заподозрить наличие у ребенка врожденной миопатии. Тщательное исследование мышц с проведением силового тестирования (эргометрии), стандартной и стимуляционной электромиографии, миотонометрии или электротонометрии позволяет получить дополнительные данные, свидетельствующие в пользу диагноза «врожденная миопатия». Исключить генерализованную воспалительную миопатию (дерматомиозит, полимиозит) и диффузный миозит позволяет отсутствие болевого синдрома, уплотнений и воспалительной отечности мышц.
Окончательно врожденная миопатия может быть диагностирована только после результатов морфологического исследования мышечной ткани, полученной путем биопсии мышц. Лишь это обследование позволяет определить специфичные для каждого вида миопатии изменения и установить точный диагноз. Однако даже биопсия не всегда позволяет достоверно верифицировать тип врожденной миопатии.
Лечение врожденной миопатии
К сожалению, на сегодняшний день не существует достаточно эффективных способов лечения и врожденная миопатия сохраняет свои проявления в течение всей жизни больного. Возможные методы терапии направлены на поддержание как можно более высокого уровня жизнеспособности пациента. Если врожденная миопатия сопровождается значительным снижением мышечной силы, в первые месяцы жизни медицинские мероприятия заключаются в борьбе с дыхательной недостаточностью, обеспечении питания через желудочный зонд, купировании бронхо-легочных осложнений. Благоприятное влияние на состояние больных в любом возрасте оказывает массаж, водолечение и физиотерапевтические процедуры. В более старшем возрасте может потребоваться ортопедическая коррекция имеющихся нарушений и социальная адаптация пациентов.
www.krasotaimedicina.ru
Миотубулярная миопатия — Мед Др
Этот тип врожденной миопатии описан в 1966 г. A. Spiro с соавт. К настоящему времени накопилось довольно значительное количество публикаций, отличающихся как фенотипическими проявлениями страдания, так и типом наследования. Отмечены аутосомно-доминантный, аутосомно-рецессивный и рецессивный, сцепленный с Х-хромосомой типы наследования. Эти обстоятельства позволяют говорить о генетической гетерогенности миотубулярной миопатии.
При описании клиники авторы подчеркивают генерализованный характер мышечной гипотонии и слабости с участием нередко глазодвигательных мышц (двусторонний умеренный птоз, парез наружных мышц глаза). Могут иметь место умеренные костные деформации. Течение заболевания различное, чаще это стационарный дефект или симптомы постепенно регрессируют, однако описаны случаи со слабым прогрессированием. В большинстве случаев клинические симптомы миотубулярной миопатии отмечаются с первых дней жизни, но иногда они проявляются впервые у взрослых.
Электромиография выявляет обычно мышечный характер поражения, но нередко отмечается типичный спинальный тип электрической активности с высокой амплитудой биопотенциалов и урежением частоты. Иногда ЭМГ имеет смешанный тип.
Патогномонична для данного типа миопатии картина морфологических изменений. Световая микроскопия часто выявляет уменьшение диаметра мышечных волокон. В центре этих малых волокон миофибриллы отсутствуют или очень рыхло упакованы.
Гистохимические реакции выявляют в центральных областях высокую активность окислительных ферментов, высокое содержание гликогена, митохондриальной АТФ-азы и α-глюканфосфорилазы.
По периферии таких ненормальных мышечных волокон миофибриллы могут иметь нормальный вид с низкой активностью ферментов и нормальным содержанием гликогена. По данным электронной микроскопии парануклеарная зона содержит участки саркоплазмы с большим количеством митохондрий и миелиновыми фигурами.
Измененные мышечные волокна с повышенной ферментативной активностью в центре очень напоминают эмбриональные мышечные клетки или миотубулы. В связи с этим заболевание и получило название миотубулярной миопатии.
Другое ее название — центронуклеарная миопатия, что обусловлено скоплением в центральной части мышечного волокна группы ядер. Имеется предположение о том, что формирование миотубул связано с дефектом иннервации на эмбриональном уровне, предложен даже термин «миотубулярная нейропатия».
Однако результаты тщательного посмертного патоморфологического исследования, а также изучение терминальных нервных окончаний при витальном окрашивании не подтвердили патологии последних. Представляет интерес мнение Radu Н. и соавт. (1977), которые предполагают раннюю функциональную денервацию с нарушением аксоплазматического тока по нерву.
«Нервно-мышечные болезни»,
Б.М.Гехт, Н.А.Ильина
Читайте далее:
www.meddr.ru
причины, симптомы, диагностика и лечение
Врожденные миопатии — это широкое понятие нервных мышечных расстройств, группа редких первичных дефектов мышц, которые передаются по наследству, вызывают гипотонию при рождении или в период новорожденности. Заболевания, относящиеся к группе врожденных миопатий, имеют сложную клиническую картину и схожие симптомы, что значительно усложняет их диагностирование и лечение.
Содержание статьи:
Проявляется как диффузная мышечная слабость и снижение мышечного тонуса, степень выраженности которых напрямую зависит от разновидности миопатии и уровня ее сложности. В тяжелых случаях может привести к летальному исходу от дыхательной недостаточности.
Врождённая миопатия — заболевание генетически обусловленное. Различные виды миопатии могут находиться в разных локусах хромосом, поэтому могут передаваться по наследству рецессивно, доминантно или совместно с Х-хромосомой. Генетическая патология нарушает синтез белка, который входит в состав мышечной ткани, что приводит к нарушению строения мышечных волокон. В следствие этого, мышцы утрачивают возможность нормально сокращаться, наблюдается мышечная слабость.
Проявляется врожденная миопатия обычно в раннем детском возрасте (очень редко проявляется у взрослых) и сохраняет свою симптоматику на протяжении всей жизни больного. Чаще всегоданное заболевание слабо прогрессирует или не прогрессирует совсем.
Патогенез миопатии до конца не изучен и причины появления не выяснены. Существует гипотеза «дефекта мембран» мышечных клеток и цитоплазматических органелл, которую многие неврологи называют причиной зарождения заболевания.
Классификация врожденной миопатии
Биопсия мышц дает определенные биохимические и морфологические данные, на основании которых все заболевания врожденной миопатии можно условно разделить две большие группы.
Врожденные мышечные дистрофии
Первая группа — это врожденные мышечные дистрофии (нарушение функции и строения мышечных волокон в результате сбоя синтеза белков, которые входят в их состав). Четкой классификации врожденных мышечных дистрофий нет, но на основании гипотез патогенеза данной группы заболевания можно выделить две группы:
- мерозин-негативные заболевания, которые характеризуются дефицитом или отсутствием белка мезонина, входящего в состав поперечно-полосатых мышц;
- врожденные структурные миопатии и мерозин-позитивные, при которых концентрация мезонина находится в норме.
Мерозин-негативная группа заболеваний подразделяется на несколько типов:
- врожденные мышечные дистрофии Фукуямы;
- мышечно-глазо-мозговой синдром;
- синдром Уолкера-Варбурга.
Клиническая картина заболеваний мерозин-негативной группы очень схожа с симптоматикой классических врожденных миопатий, но отличительной особенностью является вовлечение в общую симптоматику различных структур головного мозга, что ведет к дальнейшей умственной отсталости, задержке развития. Заболевания мерозин-позитивной группы намного реже включают в себя поражение центральной нервной системы (приблизительно у 10% больных выявлено поражение мозга) и обычно не влечет за собой торможение интеллекта. Клиническая картина характеризуется деформацией позвоночника и нарушением черт строения лица.
Врожденные структурные миопатии
Вторая группа — врожденные структурные миопатии (нарушения целостности цитоскелета мышечных волокон и возникновение патологии в биоптате мышц). Эта группа заболеваний характеризуется нарушением синтеза белков, отвечающих за рост и другие функции формирования мышц в раннем развитии эмбриона.
К врожденным структурным миопатиям относят:
- болезнь центрального стержня;
- немалиновую миопатию;
- центронуклеарную миопатию;
- мегакониальную миопатию;
- миопатию с диспропорцией типов мышечных волокон;
- миопатию со множественными центральными стержнями;
- миотубулярную миопатию;
- миопатию с кристаллическими включениями.
Клинические картины каждого из заболеваний данной группы похожи друг на друга и характеризуются мышечной гипотонией и гипертрофией, пониженной рефлективностью в сухожильях и повышением концентрации в крови креатинфосфокиназы. Наблюдается медленное прогрессирование.
Симптомы врожденной миопатии
Врожденная миопатия дебютирует чаще всего в первые месяцы жизни ребенка. Характеризуются данные заболевания наличием синдрома «вялого ребенка»: заметное снижение мышечного тонуса, слабость в мышцах, плохо развивается мускулатура и наблюдается обессиливание во время процесса сосания. С развитием ребенка мышечная слабость более заметно выражена — нехватка сил для того, чтобы стать на ножки или просто поднять свое тело, могут возникнуть трудности при ходьбе или сидении, наблюдается заметное отставание в физическом развитии по сравнению с другими детьми такого же возраста.
Слабость в мышцах может быть выражена сильно или незначительно. Чаще всего симптоматика сохраняется на весь период жизни больного и практически не прогрессирует или слабо развивается. В отдельных случаях, можно наблюдать невозможность самостоятельно передвигаться, поэтому больной вынужден использовать коляску, но навыки самообслуживания, приобретенные им, не утрачиваются.
Врожденные миопатии провоцируют не только слабость мышц конечностей и спины, слабеют и мышцы дыхательной мускулатуры, что является особенно опасным для детей грудного возраста. Если мышечная слабость дыхательных путей выражена в малой степени, то наблюдается развитие дыхательной недостаточности. Это, в свою очередь, провоцирует различные заболевания дыхательных путей (бронхиты, всевозможные виды пневмоний). Иногда дыхательная недостаточность приводит к летальному исходу еще в младенческом возрасте. Бывают случаи, когда с возрастом слабость мышц уменьшается или наоборот прогрессирует.
В отдельных случаях врожденная миопатия проявляется также в виде дисморфичных черт лица (удлиненная форма черепа, высокое небо) или патологиями развития скелета (сколиоз, косолапость, врожденный вывих бедра, кифоз).
Характеристика отдельных видов врожденной миопатии
Болезнь центрального стержня
Наследуется аутосомно-доминантным способом с неполной пенетрантностью (но встречаются и спорадические случаи наследственности). Данная форма врожденной миопатии характеризуется патологией проксимальных мышц конечностей, но больные способны приобрести некоторые двигательные навыки. В младенческом возрасте наблюдается задержка двигательного развития и гипотония, но диагностировать данное заболевание можно только в более позднем возрасте при изменениях скелета и выраженной мышечной слабости. При этом наблюдаются патологии скелета: деформация стоп, кифосколиоз, дислокация бедер, грудь сапожника.
Чаще всего, больные имеют хрупкую фигуру и невысокий рост. При диагностике заболевания проводят биоптат мышц, который показывает наличие множественных или единичных прерывистых зон, которые лишены ферментов окисления, в некоторых мышечных волокнах. Проведение других лабораторных анализов может показать норму. Пациенты с болезнью центрального стержня склонны к развитию злокачественной гипертермии.
Немалиновая миопатия
Второе название данного заболевания — врожденная непрогрессирующая нитеобразная миопатия. Наследственность в основном передается по аутосомно-доминантному типу, но встречается также рецессивный и спорадический. Возможен летальный исход вследствие дыхательной недостаточности в раннем младенческом возрасте. Наблюдается сильно выраженные патологии скелета. Развитие болезни может происходить в той или иной степени, а может не прогрессировать вовсе. В отдельных случаях больные вынуждены передвигаться с помощью сидячей каталки, в других — страдают от дыхательной недостаточности. При диагностировании проводится гистологическое исследование, которое выявляет в мышцах неподобные или палочкоподобныене малиновые тельца. ЭМГ обычно утверждает диагноз миопатии.
Миотубулярная миопатия
Данный тип врожденной миопатии наследуется по аутосомно-рецессивному типу. Миотубулярная миопатия характерна наличием центрально расположенных ядер в большинстве мышечных волокон. Это напоминает вид мышцы на миотубулярном внутриутробном развитии плода. В следствии этого заболевание и получило свое название.
Диагностика врожденной миопатии
Диагностика при врожденных миопатиях — сложный процесс, поскольку врачу необходимо дифференцировать и определить конкретный вид миопатии для назначения адекватного лечения и постановки правильного диагноза. Врач-невропатолог выявляет неврологические симптомы, проводит электрофизиологическое и биохимическое исследование, чтобы установить гетерозиготное носительство миопатического гена. Электромиографическое исследование с помощью накожных электродов показывает зачастую снижение вольтажа кривой ЭМГ. При биохимическом анализе крови в сыворотке наблюдается повышенная концентрация альдолазы и креатинкиназы.
Лечение врожденной миопатии
Лечение врожденный миопатий малоэффективное. Четкого лечения на данный момент нет. О том, можно ли лечить врожденную миопатию, ученые спорят до сих пор. Медицинские институты разных стран проводят исследования на генном уровне — с использованием стволовых клеток. Существует симптоматическое лечение, которое заключается в воздействии на обменные процессы в организме пациента, в частности, синтез белков, попытку нормализации функций вегетативной нервной системы. Чаще всего медикаментозное лечение включает в себя принятие анаболических гормонов (неробол, цераксон, ретаболил, сомазин), АТФ. Обязательно проводится витаминотерапия. Назначаются также антихолинэстеразные препараты.
Обязательным звеном процесса лечения врожденных миопатий является лечебная физкультура. Это могут быть занятия в воде или комплекс упражнений. ЛФК контролируется тренером или неврологом. В отдельных случаях действенной оказывается ортопедическая коррекция (например, ношение ортопедической обуви, корсетов или использование ортопедических матрасов, подушек, кресел).
Состояние и клиническую картину заболевания контролирует невропатолог, терапевт, педиатр, кардиолог и ортопед-травматолог.
www.mosmedportal.ru
Центр ветеринарной генетики ЗООГЕН — Миотубулярная миопатия
Myotubular myopathy 1, X-linked myotubular myopathy, MTM1, XL-MTMОписание
Описание
Нервно-мышечное заболевание, схожее симптомами с центроядерной миопатией. У больных животных существенно снижен уровень синтеза миотубулярина, – фосфатазы, участвующей в контроле процессов роста, пролиферации и дифференцировки клеток. Дефекты этого белка сопровождаются нарушениями нормального процесса созревания мышц.
Заболевание сцеплено с X-хромосомой, в связи с чем преимущественно встречается у кобелей. Появление больной суки возможно только в том случае, если оба родителя передали по копии мутантного аллеля, но так как кобелей обычно подвергают эвтаназии в раннем возрасте, они не успевают дать потомство.
Симптоматика
- Первые симптомы проявляются у щенков в возрасте между 7 и 19 неделями. Признаки заболевания включают в себя скелетную мышечную атрофию, мышечную слабость, задержку в росте, проблемы с питанием из-за слабости в жевательных мышцах, хриплый лай, внезапное падение при ходьбе. Заболевание прогрессирует достаточно быстро, и щенки теряют способность стоять и поднимать голову через 3-4 недели после появления первых симптомов.
Интерпретация результатов
Сцепленный с X-хромосомой рецессивный характер наследования
Сука
MM — болен
NM — здоров, носитель
NN — здоров
Кобель
MY — болен
NY — здоров
Молекулярная генетика (для специалистов)
- Причиной заболевания у лабрадоров ретриверов является однонуклеотидная замена c.465C>A (p.N155K) в 7-м экзоне гена МТМ1 (Beggs et al., 2010).
Литературные данные
- Beggs, A.H., Böhm, J., Snead, E., Kozlowski, M., Maurer, M., Minor, K., Childers, M.K., Taylor, S.M., Hitte, C., Mickelson, J.R., Guo, L.T., Mizisin, A.P., Buj-Bello, A., Tiret, L., Laporte, J., Shelton, G.D.: MTM1 mutation associated with X-linked myotubular myopathy in Labrador Retrievers. Proc Natl Acad Sci U S A 107:14697-702, 2010. Pubmed reference: 20682747.
zoogen.org
причины, симптомы, диагностика и лечение
Миопатии — группа заболеваний, основу которых составляют различные нарушения в метаболизме и строении мышечной ткани, приводящие к снижению силы пораженных мышц и ограничению двигательной активности. Типичными чертами миопатии являются: прогрессирующая мышечная слабость, развитие мышечных атрофий, снижение сухожильных рефлексов и тонуса мышц. Установить диагноз миопатии помогают электрофизиологические исследования, биохимические анализы крови и мочи, результаты молекулярно-генетического и гистохимического анализа образцов, полученных путем биопсии мышц. Лечение предполагает комплексное назначение метаболических препаратов курсами 3 раза в год.
Общие сведения
Миопатии относятся к группе нервно-мышечных заболеваний. Характеризуются дистрофическим поражением мышечной ткани (преимущественно скелетной мускулатуры) с выборочной атрофией отдельных волокон (миофибрилл) при полной функциональной сохранности анимальной нервной системы. Отличаются хроническим неуклонно прогрессирующим течением. Как правило, манифестация клинических проявлений миопатии приходится на детский и юношеский возраст. Большую часть случаев заболевания представляет генетическая патология — это так называемые первичные миопатии. Реже встречаются миопатии приобретенного генеза — вторичные или симптоматические.
Миопатии
Причины миопатий
В основе первичных миопатий лежат генетически детерминированные нарушения в функционировании митохондрий и ионных каналов миофибрилл, в синтезе мышечных белков или ферментов, регулирующих обмен веществ мышечной ткани. Наследование дефектного гена может происходить рецессивно, доминантно и сцеплено с Х-хромосомой. При этом внешние факторы зачастую выступают в роли триггеров, запускающих развитие болезни. Подобными «пусковыми» факторами могут являться разнообразные инфекции (хронический тонзиллит, частые ОРВИ, бактериальная пневмония, сальмонеллез, пиелонефрит и пр.), алиментарная дистрофия, тяжелые травмы (перелом костей таза, политравма, ЧМТ и др.), физическое перенапряжение, интоксикации.
Приобретенные миопатии могут развиваться на фоне эндокринных расстройств (гиперпаратиреоза, болезни Иценко-Кушинга, гиперальдостеронизма), хронических интоксикаций (токсикомании, наркомании, алкоголизма, профессиональных вредностей), мальабсорбции и авитаминозов, тяжелых хронических заболеваний (ХПН, хронической печеночной недостаточности, сердечной недостаточности, ХОБЛ), опухолевых процессов.
Патогенез
Наличие генетически детерминированных или приобретенных дефектов метаболитов, участвующих в обмене веществ и построении мышечных волокон, приводит к возникновению и прогрессированию дегенеративных изменений последних. Развивается атрофия миофибрилл, происходит их замещение жировой и соединительной тканью. Мышцы утрачивают способность к сокращению, что обуславливает мышечную слабость и ограничение возможности выполнять активные движения.
Последние исследования выявили у больных различными формами миопатий нарушения функционирования как центральных (на диэнцефальном уровне), так и периферических отделов вегетативной нервной системы, играющих не последнюю роль в патогенезе заболевания. Именно этим можно объяснить типичное для миопатий преимущественное поражение проксимальных отделов конечностей, имеющих более богатую вегетативную иннервацию.
Классификация
Специалистами в области неврологии разработано несколько классификаций миопатий. Наибольшую популярность среди клиницистов получил этиопатогенетический принцип разделения, согласно которому выделяют наследственные, воспалительные, метаболические, мембранные, паранеопластические и токсические миопатии. Среди наследственных миопатий наиболее распространены 3 вида: ювенильная/юношеская форма Эрба, псевдогипертрофическая форма Дюшена и плече-лопаточно-лицевая форма. Реже встречаются скапулоперонеальная, окулофарингеальная, дистальная и др. формы. Отдельной группой идут врожденные миопатии: болезнь центрального стержня, немалиновая и миотубулярная миопатия, диспропорция типов миофибрилл.
Воспалительные миопатии классифицируются как инфекционные — возникающие вследствие инфекционно-воспалительного поражения мышечной ткани при различных инфекционных процессах: бактериальных (стрептококковая инфекция), вирусных (энтеровирусы, грипп, краснуха, ВИЧ), паразитарных (трихинеллез, токсоплазмоз) и идиопатические — дерматомиозит, миозит с включениями, полимиозит, миопатии при коллагенозах.
Метаболические миопатии подразделяются на связанные с нарушением липидного обмена в мышцах (недостаточность ацетил-КоА-дегидрогеназы, дефицит карнитина), обмена гликогена (болезнь Андерсена, болезнь Помпе, гликогеноз III типа, болезнь Мак-Ардля, дефицит киназы фосфорилазы b, дефицит фосфоглицеромутазы), метаболизма пуринов (дефицит фермента МАДА) и митохондриальные миопатии (дефицит редуктазы, АТФ, цитохрома b, b1).
Симптомы миопатий
Большинство миопатий имеют постепенное начало с появления небольшой мышечной слабости в конечностях, более быстро возникающей усталости от ходьбы и другой физической нагрузки. В течение нескольких лет происходит нарастание слабости, появляются и прогрессируют мышечные атрофии, возникают деформации конечностей. Из-за значительной мышечной слабости пациенты с трудом поднимаются с пола и ходят по лестнице, не могут прыгать и бегать. Для того, чтобы встать со стула, им приходится использовать специальные приемы. Характерен вид больного: крыловидно отстоящие лопатки, опущенные плечи, выпяченный вперед живот и усиленный поясничный лордоз. Наблюдается «утиная» походка — пациент передвигается, раскачиваясь в стороны.
Патологические изменения при миопатиях происходят симметрично в мышцах конечностей и туловища. Как правило, мышечные атрофии наблюдаются в проксимальных отделах рук и ног. В связи с этим мышцы дистальных отделов конечностей могут выглядеть гипертрофированными. Такая миопатическая псевдогипертрофия наиболее заметна в мышцах голеней. Наряду с нарастанием мышечной слабости наблюдается постепенное угасание сухожильных рефлексов и прогрессирующее снижение мышечного тонуса, т. е. развивается и усугубляется периферический вялый паралич. Со временем результатом резкого ограничения активных движений становятся контрактуры суставов.
Миопатии могут сопровождаться поражением мимических мышц, что проявляется невозможностью вытянуть губы трубочкой, свистеть, нахмурить лоб или улыбнуться. Поражение круговой мышцы рта приводит к появлению дизартрии, связанной с затруднением произношения гласных звуков.
Клиника некоторых миопатий включает поражение дыхательной мускулатуры, приводящее к возникновению застойной пневмонии и развитию дыхательной недостаточности. Возможны патологические изменения сердечной мышцы с возникновением кардиомиопатии и сердечной недостаточности, мышц глотки и гортани с развитием дисфагии и миопатического пареза гортани.
Особенности отдельных форм миопатии
Ювенильная миопатия Эрба наследуется аутосомно-рецессивно. Патологические процессы начинают проявляться в возрасте 20-30 лет. В первую очередь они охватывают мышцы тазового пояса и бедер, затем быстро распространяются на другие мышечные группы. Вовлечение лицевой мускулатуры не характерно. Начало миопатии в более молодом возрасте приводит к ранней обездвиженности пациентов. При развитии заболевания в старшем возрасте его течение менее тяжелое: пациенты длительно сохраняют способность передвигаться.
Псевдогипертрофическая миопатия Дюшена наследуется рецессивно сцеплено с полом. Болеют исключительно мальчики. Как правило, манифестирует в течение первых 3-х лет жизни, реже — в период от 5 до 10 лет. Типично начало с атрофических изменений мышц тазового пояса и проксимальных отделов ног, сопровождающихся псевдогипертрофией икроножных мышц. Рано возникают контрактуры и искривление позвоночника (кифоз, сколиоз, гиперлордоз). Может наблюдаться олигофрения. Заболевание протекает с поражением дыхательных мышц и сердца (кардиомиопатия отмечается у 90% больных миопатией Дюшена), что является причиной раннего летального исхода.
Плече-лопаточно-лицевая миопатия Ландузи – Дежерина имеет аутосомно-доминантное наследование. Манифестирует в 10-20 лет с поражения мимических мышц. Постепенно слабость и атрофии охватывают мышцы надплечий, плеч и груди. Мышцы тазового пояса обычно не страдают. Характерно медленное течение с длительной сохранностью работоспособности, без сокращения продолжительности жизни.
Скапулоперонеальная миопатия — аутосомно-доминантное заболевание. Его особенностью является развитие атрофий в мышцах дистальных отделов ног и проксимальных отделов рук, а также наличие легких сенсорных нарушений дистальных отделов как нижних, так и верхних конечностей.
Окулофарингеальная миопатия характеризуется сочетанием поражения глазодвигательных мышц со слабостью мышц языка и глотки. Обычно манифестирует двусторонним птозом, затем присоединяются расстройства глотания. Особенностью этой миопатии является ее позднее начало — на 4-6-ом десятилетии жизни.
Дистальная поздняя миопатия наследуется аутосомно-доминантно. Отличается развитием слабости и атрофий в дистальных отделах конечностей: вначале в стопах и кистях, а затем в голенях и предплечьях. Характерно медленное течение.
Особенности клинических проявлений различных форм врожденных, наследственных и метаболических миопатий описаны в самостоятельных обзорах.
Диагностика
Установить диагноз миопатии неврологу помогают электрофизиологические методы обследования: электронейрография (ЭНГ) и электромиография (ЭМГ). Они позволяют исключить поражение периферического двигательного нейрона и, таким образом, дифференцировать миопатию от инфекционной миелопатии, нарушений спинномозгового кровообращения, миелита и опухолей спинного мозга. Данные ЭМГ говорят о характерных для миопатий изменениях мышечных потенциалов — уменьшении их амплитуды и сокращении длительности. О прогрессирующем процессе свидетельствует наличие большого количества коротких пиков.
Биохимический анализ крови при миопатии показывает повышение содержания альдолазы, КФК, АЛТ, АСТ, ЛДГ и др. ферментов. В биохимическом анализе мочи показательным является увеличение концентрации креатинина. В установлении формы миопатии первостепенное значение имеет биопсия мышц. Морфологическое исследование образцов мышечной ткани выявляет наличие беспорядочно разбросанных атрофированных миофибрилл среди практически сохранных и гипертрофированных мышечных волокон, а также замещение участков мышечной ткани на соединительную или жировую. Постановка окончательного диагноза возможна только после сопоставления результатов гистохимических, иммунобиохимических и молекулярно-генетических исследований.
С целью диагностики поражений сердечной мышцы пациенту с миопатией могут быть назначены консультация кардиолога, ЭКГ, УЗИ сердца; при подозрении на возникновение пневмонии — консультация пульмонолога и рентгенография легких.
Лечение миопатий
В настоящее время патогенетическое лечение миопатий находится в состоянии научных экспериментов в области генной инженерии. В клинической практике применяется симптоматическая терапия, состоящая в основном в улучшении метаболизма мышечной ткани. С этой целью применяют витамины Е, В1, В6, В12, АТФ, неостигмин, аминокислоты (глютаминовую кислоту, гидролизат из мозга свиньи), антихолинэстеразные препараты (амбеноний, галантамин), анаболические стероиды (нандролона деканоат, метандиенон), препараты калия и кальция, тиаминпирофосфат. Комбинации из нескольких препаратов назначают курсом 1-1,5 мес. 3 раза в год.
Медикаментозное лечение миопатий дополняют физиотерапией (электрофорез с неостигмином, ионофорез с кальцием, ультразвук), легким массажем и ЛФК. Проведение ЛФК может осуществляться в бассейне. Комплекс упражнений должен быть подобран таким образом, чтобы избежать перегрузки ослабленной мускулатуры. В некоторых случаях пациенты нуждаются в консультации ортопеда и подборе средств ортопедической коррекции (корсетов, обуви).
Основу лечения приобретенных форм миопатий составляет терапия основного заболевания: коррекция эндокринных нарушений, устранение токсического воздействия и дезинтоксикация организма, ликвидация инфекционного процесса, перевод хронического заболевания в стадию устойчивой ремиссии и т. д.
Прогноз и профилактика
Наиболее неблагоприятны в прогностическом плане наследственные миопатии, проявляющиеся в раннем детском возрасте. В остальном прогноз зависит от формы миопатии, вовлеченности в процесс сердечной и дыхательных мышц. Прогноз вторичных миопатий более благоприятный при условии успешного лечения основного заболевания.
Профилактикой первичных миопатий служит тщательный сбор семейного анамнеза и обязательное консультирование у генетика пар, планирующих беременность. Профилактикой вторичных миопатий является исключение токсических воздействий на организм, своевременное лечение инфекционных и эндокринных заболеваний, коррекция метаболических нарушений.
www.krasotaimedicina.ru