
Синдром бар – Синдром Луи-Бар — причины, симптомы, диагностика и лечение
Синдром Луи-Бар — причины, симптомы, диагностика и лечение
Синдром Луи-Бар (атаксия-телеангиэктазия) — наследственное заболевание, проявляющееся мозжечковой атаксией, телеангиэктазиями кожи и конъюнктивы глаз, недостаточностью Т-клеточного звена иммунитета. Последнее приводит к тому, что синдром Луи-Бар сопровождается частыми респираторными инфекциями и склонностью к возникновению злокачественных опухолей. Диагностируется синдром Луи-Бар на основании анамнеза и клинической картины заболевания, данных иммунограммы, результатов офтальмологического и отоларингологического обследования, МРТ головного мозга и рентгенографии легких. В настоящее время синдром Луи-Бар не имеет специфического и эффективного лечения.
Общие сведения
Синдром Луи-Бар впервые был описан в 1941 году во Франции. Нет точных данных о том, с какой частотой синдром Луи-Бар встречается среди современного населения. По некоторым сведениям эта цифра составляет 1 случай на 40 тысяч новорожденных. Однако, необходимо учитывать, что при смерти в раннем детском возрасте синдром Луи-Бар обычно остается не диагностированным. Известно, что заболевание одинаково часто поражает мальчиков и девочек. В неврологии синдром Луи-Бар относится к так называемым факомотозам — генетически обусловленным сочетанным поражениям кожи и нервной системы. В эту группу также входят нейрофиброматоз Реклингхаузена, ангиоматоз Стерджа—Вебера, туберозный склероз и др.
Синдром Луи-Бар
Причины и патогенез синдрома Луи-Бар
В основе патологических изменений, сопровождающих синдром Луи-Бар, лежат генетические нарушения, приводящие к развитию врожденной нейроэктодермальной дисплазии. Синдром Луи-Бар является аутосомно-рецессивным заболеванием, т. е. проявляется клинически только при получении рецессивного гена сразу от обоих родителей.
Морфологически атаксия-телеангиэктазия характеризуется дегенеративными изменениями тканей мозжечка, в частности потерей зернистых клеток и клеток Пуркинье. Дегенеративные изменения могут затрагивать зубчатое ядро мозжечка (nucleus dentatus), черную субстанцию (substantia nigra) и некоторые отделы коры головного мозга, иногда поражаются спиномозжечковые пути и задние столбы спинного мозга.
Синдром Луи-Бар сочетается с гипоплазией или аплазией тимуса, а также с врожденным дефицитом IgA и IgE. Эти нарушения в системе иммунитета приводят к появлению у пациентов частых инфекционных заболеваний, склонных к длительному и осложненному течению. Кроме того, иммунные нарушения могут потенцировать развитие злокачественных новообразований, зачастую берущих свое начало в структурах лимфоретикулярной системы.
Клинические проявления синдрома Луи-Бар
Атаксия. Наиболее часто синдром Луи-Бар начинает проявляться клинически в возрасте от 5 месяцев до 3 лет. Во всех случаях заболевания синдром Луи-Бар манифестирует с появления мозжечковой атаксии, признаки которой становятся очевидными, когда ребенок начинает ходить. Наблюдаются нарушения равновесия и походки, дрожание во время двигательного акта (интенционный тремор), качание туловища и головы. Зачастую атаксия настолько выражена, что имеющий синдром Луи-Бар больной не может ходить. Мозжечковая атаксия сочетается с мозжечковой дизартрией, характеризующейся невнятной скандированной речью. Отмечается мышечная гипотония, снижение или полное исчезновение сухожильных рефлексов, нистагм, глазодвигательные нарушения и косоглазие.
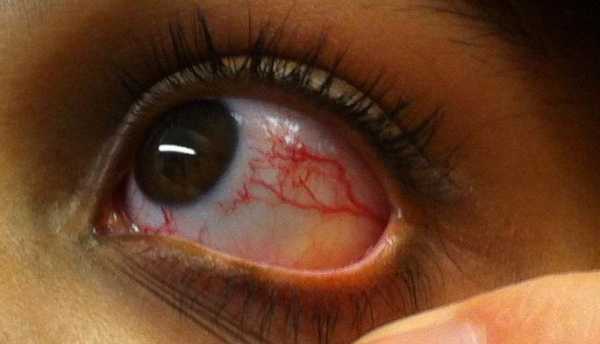
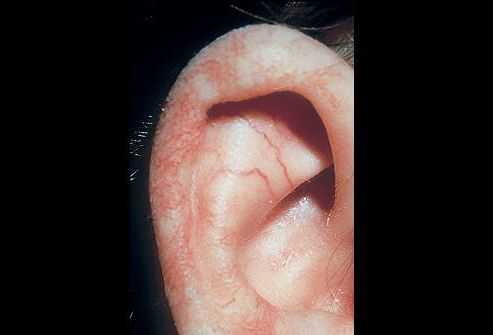
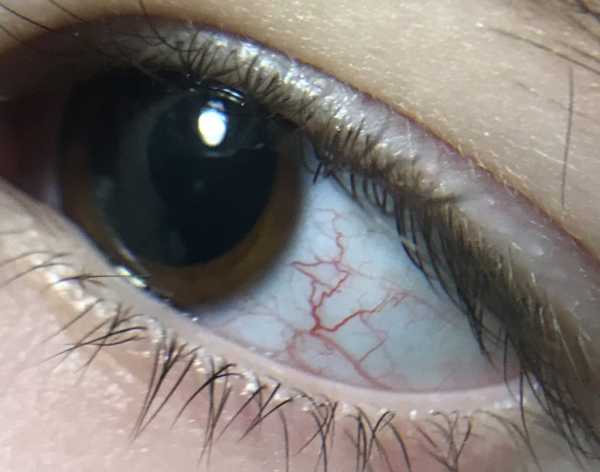
Телеангиэктазии. В большинстве случаев появление сопровождающих синдром Луи-Бар телеангиэктазий происходит в возрасте от 3 до 6 лет. В некоторых случаях их возникновение отмечается в более поздний период и очень редко в течение первого месяца жизни. Телеангиэктазии (сосудистые звездочки) представляют собой имеющие различную форму красноватые или розовые пятнышки или разветвления. Они обусловлены расширением мелких сосудов кожи. Следует отметить, что телеангиэктазии могут быть проявлением многих других заболеваний (например, розацеа, СКВ, дерматомиозита, пигментной ксеродермы, хронического лучевого дерматита, мастоцитоза и пр.). Однако в сочетании с атаксией они дают специфическую для синдрома Луи-Бар клиническую картину.
Синдром Луи-Бар характеризуется изначальным возникновением телеангиэктазий на конъюнктиве глазного яблока, где они имеют вид «паучков». Затем сосудистые звездочки появляются на коже век, носа, лица и шеи, локтевых и коленных сгибов, предплечий, тыльной поверхности стоп и кистей. Телеангиэктазии могут также наблюдаться на слизистой оболочке мягкого и твердого неба. Наиболее выражены сосудистые звездочки в тех местах кожного покрова, где он подвергается воздействию солнечных лучей. В первую очередь это лицо, где телеангиэктазии образуют целые «пучки». При этом кожа теряет свою эластичность и становится плотной, что напоминает изменения, типичные для склеродермии.
Кожные проявления атаксии-телеангиэктазии могут включать появление веснушек и пятен цвета кофе с молоком, участков обесцвеченной кожи. Наличие гипо- и гиперпигментаций делает кожные симптомы синдрома Луи-Бар схожими с клиникой пойкилодермии. У многих больных отмечается сухость кожи и участки гиперкератоза. Может наблюдаться гипертрихоз, ранняя седина волос, кожные элементы, напоминающие акне или проявления псориаза.
Инфекции дыхательных путей. Характеризующее синдром Луи-Бар поражение иммунной системы приводит к возникновению частых рецидивирующих инфекций дыхательных путей и уха: хронических ринитов, фарингитов, бронхитов, пневмоний, отитов, синуситов. Их особенностями являются: стертость границ между периодом обострения и ремиссии, скудность физикальных данных, плохая чувствительность к антибактериальной терапии и длительное течение. Каждая подобная инфекция может стать для больного атаксией-телеангиэктазией смертельно опасной. Частые заболевания легких приводят к развитию бронхоэктазов и пневмосклероза.
Злокачественные новообразования. Среди пациентов, имеющих синдром Луи-Бар, злокачественные опухолевые процессы отмечаются в 1000 раз чаще, чем в среднем у населения. Наиболее распространенными среди них являются лейкемия и лимфома. Особенностью онкопатологии в случае синдрома Луи-Бар является повышенная чувствительность пациентов к воздействию ионизирующего излучения, что полностью исключает применение лучевой терапии при их лечении.
Диагностика синдрома Луи-Бар
Постановка диагноза атаксии-телеангиэктазии требует комплексного подхода, учитывающего анамнез заболевания, его клинические проявления, данные иммунологических и инструментальных исследований, а также результаты ДНК-диагностики. Пациент с подозрением на синдром Луи-Бар должен пройти обследование не только у невролога, но и у дерматолога, отоларинголога, офтальмолога, иммунолога, пульмонолога, онколога.
Лабораторная диагностика синдрома Луи-Бар включает клинический анализ крови, в котором у 1/3 пациентов наблюдается снижение количества лимфоцитов. Обязательно проводится исследование уровня иммуноглобулинов крови, которое выявляет значительное снижение IgA и IgE, в 10-12% случаев IgG. Примерно у 40% пациентов синдром Луи-Бар сопровождается аутоиммунными реакциями, о которых свидетельствует наличие аутоантител к митохондриям, тиреоглобулину, иммуноглобулинам.
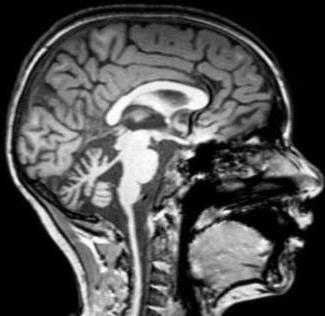
Из инструментальных способов диагностики синдрома Луи-Бар могут применяться: УЗИ тимуса, МРТ головного мозга, фарингоскопия, риноскопия, рентгенография легких. При помощи УЗИ диагностируется аплазия или гипоплазия тимуса. МРТ головного мозга выявляет атрофию мозжечка, расширение IV желудочка. Рентгенография легких необходима для диагностики очаговой или крупозной пневмонии, выявления очагов пневмосклероза и бронхоэктатических изменений.
Синдром Луи-Бар следует дифференцировать с атаксией Фридрейха, болезнью Рандю-Ослера, атаксией Пьера-Мари, болезнью Гиппеля-Линдау и др.
Лечение и прогноз синдрома Луи-Бар
К сожалению, эффективные методы лечения синдрома Луи-Бар до настоящего времени остаются предметом поиска. В современной медицине возможно применение лишь паллиативного симптоматического лечения соматических и иммунологических нарушений. Продлению жизни пациентов, имеющих синдром Луи-Бар, способствует иммунокоррегирующая терапия препаратами тимуса и гамма-глобулином, витаминотерапия в высоких дозировках и интенсивная терапия любого инфекционного процесса. По показаниям применяют противовирусные препараты, антибиотики широкого спектра действия, противогрибковые средства, глюкокортикостероиды.
В связи с отсутствием эффективных способов лечения синдром Луи-Бар имеет неблагоприятный прогноз как для выздоровления, так и для жизни. Больные этим заболеванием редко доживают до 20 лет. В большинстве случаев они погибают от инфекционных осложнений и онкологических заболеваний.
www.krasotaimedicina.ru
Синдром Луи-Бар: симптомы, диагностика и лечение
Как известно, существует много различных хромосомных аномалий, которые закладываются еще в период внутриутробного развития. Изучением этих патологий занимаются генетики. В последние годы данная область медицины активно развивается, поэтому в скором будущем такие заболевания легче будет диагностировать и лечить. К счастью, данные аномалии встречаются очень редко. Это связано с улучшением диагностики плода. Одной из патологий, связанных с хромосомными нарушениями, является синдром Луи-Бар. В большинстве случаев это заболевание выявляют в первый год жизни младенца, но иногда оно дает о себе знать лишь к 6-7 годам.

Синдром Луи-Бар – что это за патология?
Эта патология относится к врожденным генетическим порокам. В большинстве случаев она передается по наследству. Атаксия-телеангиэктазия (синдром Луи-Бар) встречается крайне редко. Данное заболевание имеет специфичные проявления, которые позволяют диагностировать эту патологию. Чтобы поставить точный диагноз, необходим консилиум врачей, которые подтвердят или опровергнут наличие страшной аномалии.
История и эпидемиология заболевания
Данный синдром встречается очень редко. Его частота составляет около 1 случая на 40 тысяч населения. Впервые заболевание было обнаружено французской женщиной-ученым Луи-Бар. Синдромы, характерные для данной патологии, она объединила в одну нозологию. Это произошло в 1941 году. После было обнаружено еще несколько случаев заболевания по всему миру. Так как данная аномалия встречается крайне редко, нельзя с точностью сказать, какова этиология синдрома Луи-Бар. Считается, что появление заболевания не зависит от климатических условий. Поэтому синдром может встречаться в любых регионах. Помимо этого, нет данных, которые связывали бы заболеваемость с полом пациента. То есть синдром Луи-Бар с одинаковой частотой наблюдается как у мальчиков, так и у девочек.
Причины развития патологии
Данная аномалия развития закладывается еще в первом триместре беременности. Заболевание передается только по наследству. Синдром относится к аутосомно-рецессивным генетическим патологиям. Это означает, что ребенок точно унаследует заболевание, если оба родителя имеют нарушение хромосом. Если же аномалия наблюдается у одного из них (независимо от пола), то шанс появления синдрома Луи-Бар у малыша составляет 50%. Основная причина мутации – это нарушение длинного плеча 11-й хромосомы. Точные факторы, которые приводят к такой генетической перестройке, неизвестны. Но выделяют ряд вредных воздействий, влияющих на эмбриональное развитие. В первую очередь это факторы окружающей среды (облучение, отравление ядовитыми веществами). Также в первом триместре беременности очень опасен стресс.

Синдром Луи-Бар: патогенез заболевания
Как и большинство врожденных хромосомных патологий, данный синдром охватывает сразу несколько органов и систем. Основными мишенями этого заболевания являются головной мозг и иммунитет человека. Также имеется и выраженное поражение кожного покрова. Все клинические проявления данного заболевания связаны с механизмом его развития. В первую очередь наблюдаются дегенеративные процессы в ЦНС. А именно, мозжечковая атаксия. При этом часть элементов не развивается (волокна Пуркинье и зернистые клетки). Другими видимыми нарушениями являются кожные проявления – телеангиэктазии. Они представляют собой расширенные сосуды, которые особенно выражены на лице (инъекция склер, ушные раковины, нос). Атаксия мозжечка и телеангиэктазии в совокупности носят название синдром Луи-Бар. Детей, рожденных с данным заболеванием, можно выделить в первые годы жизни, так как аномалия проявляется выраженными физическими нарушениями (отставание в развитии, неустойчивое положение тела, слабость мышц).

Помимо этого, патогенез заболевания включает в себя недостаточность иммунной системы (Т-лимфоцитов). У детей, страдающих данной патологией, наблюдается гипо- или полная аплазия тимуса. В результате этого клеточный иммунитет развит очень слабо и не способен обеспечивать защиту организма от инфекционных процессов.
Симптомы атаксии-телеангиэктазии
Выраженность клинической картины зависит от степени поражения мозжечка и гипоплазии вилочкой железы. Это и определяет, как будет проявляться синдром Луи-Бар. Симптомы заболевания:
- Мозжечковая атаксия. Данный синдром проявляется раньше других, обычно на первом году жизни. Он становится выражен к моменту начала самостоятельной ходьбы. Дети с атаксией мозжечка зачастую не могут стоять и нормально передвигаться. В более благоприятных случаях наблюдается шаткость походки и тремор конечностей. Помимо этого, неврологическая симптоматика выражается в слабости мышц, дизартрии различной степени (невнятная речь) и косоглазии.
- Телеангиэктазии. Кожные проявления синдрома Луи-Бар менее опасны. Обычно они дают о себе знать в возрасте от 3 до 6 лет. Телеангиэктазии – это расширенные капилляры, которые носят название «сосудистые звездочки». Больше всего они заметны на открытых участках тела, в частности на лице. Расширенные сосуды часто находятся в глазах, на носу и ушах, а также сгибательных поверхностях рук и ног.
- Склонность к инфекциям. Из-за выраженного иммунодефицита организм не справляется с вредными агентами самостоятельно. В результате у ребенка часто развиваются различные инфекции. Зачастую это хронические заболевания дыхательных путей – фарингит, ларингит, тонзиллит, пневмония.
- Опухолевые процессы. Из-за гипоплазии тимуса, помимо инфекционных процессов, организм становится восприимчив к раковым заболеваниям. Чаще всего это опухоли кроветворной и лимфоидной ткани. Если синдром Луи-Бар у ребенка является достоверным диагнозом, то ему строго запрещено лечение рака ионизирующим облучением.
Диагностика атаксии-телеангиэктазии
Диагностика синдрома Луи-Бара обычно не предоставляет большой сложности, так как его симптомы довольно специфичны. Заподозрить данное заболевание можно с первых лет жизни по клинической картине. Неврологическая симптоматика (мозжечковая атаксия, мышечная слабость, тремор и косоглазие) в совокупности с телеангиэктазиями являются показанием для диагностики этой патологии.
При подозрении на синдром Луи-Бар необходима консультация сразу нескольких специалистов. Среди них: невропатолог, дерматолог, онколог, инфекционист, эндокринолог и генетик. Помимо клинического обследования, выполняется лабораторная и инструментальная диагностика. Проводят иммунологические анализы, в которых отмечается уменьшение или полное отсутствие элементов клеточного иммунитета (снижение Т-лимфоцитов, иммуноглобулинов А, G). В ОАК наблюдается лейкоцитоз и ускорение СОЭ, что говорит о воспалительном процессе в организме. Также важна и инструментальная диагностика. Выполняется рентгенография грудной клетки (уменьшение размеров тимуса), МРТ головного мозга (дегенеративные процессы). В настоящее время, помимо стандартных исследований, проводят генетическое (исследуют нарушение 11-й хромосомы), на основании которого ставится точный диагноз.
Лечение синдрома Луи-Бар
К сожалению, этиологического лечения хромосомных аномалий на данный момент не разработано. Поэтому при данной патологии проводят лишь симптоматическую терапию и постоянное наблюдение за больным. В первую очередь лечение направлено на улучшение работы иммунной системы. Это необходимо, чтобы избежать инфекций и опухолевых процессов. С данной целью используют гамма-глобулин и препарат «Т-активин». При развитии воспалительных заболеваний применяют антибактериальные и противовирусные средства. К сожалению, синдром мозжечковой атаксии не поддается полному лечению. Чтобы приостановить дегенеративные процессы, используют ноотропные препараты. При онкологических заболеваниях прибегают к химиотерапии и хирургическому лечению.
Прогноз для жизни при синдроме Луи-Бар
Несмотря на тяжесть заболевания, при своевременной диагностике и лечении можно продлить и облегчить жизнь ребенка. С этой целью разработана паллиативная терапия для таких пациентов. К сожалению, аномалия Луи-Бар может прогрессировать быстро. В этом случае продолжительность жизни составляет 2-3 года. Иногда заболевание не развивается в течение нескольких лет. При этом продолжительность жизни значительно увеличивается. Максимальным возрастом пациентов считается 20-30 лет. В большинстве случаев причинами смерти являются инфекционные и опухолевые процессы, иногда – неврологические нарушения.
Профилактика синдрома Луи-Бар
Чтобы избежать развития данной патологии, необходимо проводить генетическое обследование плода еще на ранних сроках беременности. Также важно знать анамнез не только родителей будущего ребенка, но и других членов семьи. Во время беременности нужно избегать вредного воздействия окружающей среды и психоэмоциональных стрессов.
Если малыш с такой аномалией уже родился, то важно выполнять все назначения врача, защитить ребенка от инфекционных агентов. При слабом иммунитете и нарушенном физическом развитии необходимо своевременно диагностировать синдром Луи-Бар. Фото детей с данным заболеванием можно увидеть в специальной медицинской литературе.
fb.ru
Синдром Луи-Бар: симптомы, признаки, диагностика и лечение
Описание
Синдром Луи Бар в медицинской практике встречается не так часто, но, тем не менее, современные медики особенно опасаются данного заболевания. Это наследственная болезнь, связанная с иммунодефицитом, которая распространяется исключительно по аутосомно-рецессивному типу. В ходе патологического процесса преобладает одно из двух поражений иммунной системы, в частности страдает клеточный иммунитет. Такие потери в организме невосполнимы, а обеспечить пациенту полноценную жизнь порой просто нереально.

Рассуждая о патогенезе синдрома Луи Бар, стоит отметить, что пациентам с таким диагнозом свойственно отсутствие тимуса, а также недоразвитость лимфатических узлов и селезенки. Кроме того, до конца не сформированы органы переферии иммунной системы, вызывающие, тем самым, патогенное воздействие на человеческий ресурс со стороны различных микроорганизмов.
Причина данной патологии очевидна – генетический дисбаланс, на фоне которого еще во внутриутробном периоде преобладает нейроэктодермальная дисплазия. Имея аутосомно-рецессивное происхождение, характерный недуг передается в случае получения рецессивного гена от обоих родителей сразу.
На фоне такой аномалии прогрессируют дегенеративные изменения мозжечка, которые непосредственно затрагивают его зубчатое ядро, черную субстанцию и определенные «звенья» коры головного мозга. Такой обширный радиус действия просто не может не отразиться на генетическом и молекулярном уровне, а новорожденный появляется на свет со страшным диагнозом.
В этиологии синдрома Луи Бара также преобладает врожденный дефицит IgA и IgE, что влечет за собой учащение инфицирования организма и продолжительное лечение преобладающих заболеваний. Нарушенный на генетическом уровне иммунитет также чреват формированием злокачественных опухолей и раковых клеток. Так что крайне важно подробная диагностика и своевременное лечение маленького пациента.
Симптомы
Как правило, симптомы синдрома Луи Бара начинают проявляться в возрасте пяти месяцев – трех лет, но особенно заметны отклонения, когда малыш начинает самостоятельно передвигаться пусть и не на дальние расстояния.
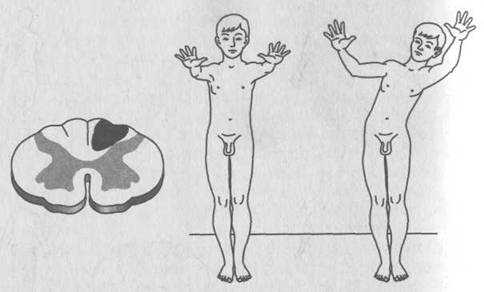
Так, признаки атаксии на лицо: шаткая и неуверенная походка, нарушенная координация движений, тремор конечностей, качание туловища и частое подергивание головы. Характерные признаки в пораженном организме зачастую настолько очевидны, что пациент просто не способен самостоятельно передвигаться. Кроме того, имеет место нарушенная речь, отсутствие сухожильных рефлексов, мышечная гипотония, косоглазие и прочие отклонения в структуре и функциональности глаз.
При данном заболевании очень часто прогрессируют инфекционные заболевания дыхательных путей и уха рецидивирующего характера. Это может быть ринит хронической формы, отит, фарингит, синусит, бронхит, реже – воспаление легких и пневмония. Однако важно понимать, что каждый последующий рецидив лишь ухудшает общее состояние, приближая летальный исход.
Еще одним красноречивым симптомом синдрома Луи Бара являются сосудистые звездочки, которые появляются, как правило, в 3 — 6 летнем возрасте. Они спровоцированы патогенным расширением небольших капилляров, однако могут свидетельствовать и о наличии других заболеваний.
Начинается телеангиэктазия на глазном яблоке виде тривиального конъюнктивита, однако уже очень скоро характерный визуальный дефект преобладает на коже век, шеи, носа, лица, локтях и тыльной стороны кисти. Также преобладает повышенная сухость кожных покровов, гиперемия, раннее выпадение волос и увеличение числа сосудистых сеточек на кожных покровах.
Синдром Луи Бара может сопровождаться появлением злокачественных новообразований, представленных лимфомой и лейкемией. Однако клинику данных патологических процессов желательно изучать в индивидуальном порядке.
Диагностика
Если у участкового терапевта возникло подозрение на присутствие синдрома Луи Бара, то он направляет его к узкому специалисту. Однако консультации у иммунолога вовсе недостаточно, ведь также стоит показаться со своей проблемой неврологу, дерматологу, офтальмологу, пульмонологу, онкологу и отоларингологу. СКрайне важно дефференцировать синдром Луи-Бара с болезнью Рандю-Ослера, атакой Фридрейха, атаксией Пьера-Мари и, конечно, мало изученным синдромом Гиппеля-Линдау.

Ставить окончательный диагноз будет невролог, однако без подробной диагностики сделать это нереально. Именно поэтому обязательно необходимо пройти инструментальное и лабораторное исследование для получения развернутой клинической картины.
Самые востребованные методы обследования представлены ниже:
- в общем анализе крови можно наблюдать патологическое снижение количества лимфоцитов;
- определение уровня иммуноглобулинов крови позволяет выявить снижение IgA и IgЕ, а также достоверно определить присутствие аутоантител к митохондриям, иммуноглобулину и тиреоглобулину;
- УЗИ помогает охарактеризовать аплазию и гипоплазию тимуса;
- МРТ головного мозга диагностировать деградацию мозжечка и патогенное расширение IV желудочка;
- Рентгенография определяет присутствие пневмонии, очагов пневмосклероза, а также преобладание бронхоэктатических изменений.
Когда все результаты диагностика, а также предварительное заключение узких специалистов будут у невролога на руках, он наконец-то определиться с окончательным диагнозом и назначит определенную схему лечения.
Профилактика
Профилактические меры не отличаются особой эффективностью, поскольку патологической процесс преобладает при непосредственном формировании эмбриона во внутриутробном периоде.
Болезнь передается по наследству и преобладает на генетическом уровне, поэтому оградить своего будущего ребенка от страшного рока весьма проблематично.
Врачи при выявлении характерной проблемы на одном из скринингов во время беременности, предлагают будущей мамочке преждевременно стимулировать роды.
Лечение
В современной медицине так и не обнаружена панацея от данного заболевания, да что там говорить, медики не могут даже определиться с общей схемой лечения. Однако в данной клинической картине однозначно требуется комплексный подход.
- Необходим продолжительный курс антибактериальной терапии, который позволяет в кратчайшие сроки истребить вторичные бактериальные инфекции, как основную причину иммунодефицита.
- Наряду с приемом антибиотиков требуется и курс гамма-глобулинов, иммуностиммуляторов, поливитаминных комплексов и даже БАДов для всеобщего укрепления ослабленного человеческого ресурса.
- В детском возрасте обязательна физиотерапия, представленная индивидуальными занятиями с логопедом по постановке речи.
Однако, так или иначе, терапия должна базироваться на основном заболевании. Если это сахарный диабет, то в схеме лечения не обойтись без пероральных сахароснижающих препаратов и инсулина. Если имеет место стремительно прогрессирующая опухоль, то требуется незамедлительное ее удаление хирургическим путем. Так что при лечении важно учитывать все нюансы, и тогда он будет, действительно, эффективным.
nebolet.com
проявления, выбор диагностики и симптоматическое лечение
Синдром Луи-Бар, известный также как атаксия-телеангиэктазия, – врожденная патология, имеющая генетическую природу. Нарушения формируются еще на раннем этапе развития плода и связаны с дефектом строения хромосомы. Клинические проявления заболевания в большинстве случаев специфические и позволяют поставить диагноз в короткие сроки. Дети с синдромом Луи-Бар страдают от двигательных расстройств на фоне дефектов строения мозжечка, у них диагностируется сосудистый рисунок на коже, слизистых и склере глаз. Поражается и иммунная система, что проявляется частыми инфекционными и вирусными заболеваниями. Лечение патологии на сегодняшний день не разработано, терапия носит симптоматический характер. В связи с этим прогноз при наличии недуга неблагоприятный.
Причины развития синдрома Луи-Бар
Основу заболевания представляет генетическая мутация, что обеспечивает формирование отклонений еще в первом триместре беременности. Происходит изменение строения плеча 11 хромосомы. Именно этот дефект и провоцирует развитие клинических признаков синдрома Луи-Бар у детей. При этом патология формируется в тех случаях, когда оба родителя являются носителями мутации. Это связано с тем, что заболевание имеет аутосомно-рецессивный тип наследования. Точные причины, провоцирующие развитие нарушения, на сегодняшний день неизвестны. Предположительно, спровоцировать у ребенка развитие синдрома Луи-Барра может пагубное воздействие на хромосомный набор, вызванное стрессом у матери на ранних сроках беременности, а также воздействие ионизирующего излучения.

Основные проявления недуга
Главными мишенями генетической аномалии являются структуры головного мозга и иммунная система человека. Именно с их поражением связано большинство клинических признаков заболевания. Синдром Луи Барра имеет несколько основных симптомов, которые считаются патогномоничными, то есть позволяют поставить диагноз. В ряде случаев у младенцев и детей школьного возраста отмечаются и другие проявления патологии, встречающиеся не так часто.
Мозжечковая атаксия
В результате генетической мутации нарушается процесс закладки нервной трубки. Это сопровождается дефектами различных отделов головного мозга. Наиболее выраженным изменениям подвержены мозжечок, некоторые участки коры и черная субстанция. Подобные нарушения сопровождаются специфической симптоматикой. Она проявляется у ребенка в возрасте от 5 месяцев до 3–4 лет. Данная особенность связана с тем, что именно в этот период малыши начинают активно ползать и учиться ходить. У пациентов отмечается выраженная атаксия, то есть неустойчивость вплоть до полной невозможности удерживать равновесие. В ряде случаев синдром Луи-Бар сопровождается и нарушением речи, которая представляется невнятной. Данный дефект также обусловлен аномалиями развития мозжечка. В связи с такими изменениями наблюдается мышечная слабость, снижение сухожильных рефлексов.
Телеангиэктазия
Термин обозначает расширение поверхностных мелких капилляров и венул кожи, склеры и слизистых оболочек, что сопровождается формированием специфичных «узоров» и сосудистой сеточки. Данный симптом проявляется у детей, как правило, в возрасте с 3 до 6 лет, в редких случаях возникает позднее. Такое клиническое проявление свойственно и многим других заболеваний. Однако в сочетании с атаксией этот признак позволяет подтвердить наличие синдрома Луи-Бара.
Телеангиэктазия наблюдается преимущественно на лице, склере глаз, а также в области локтевых и коленных сгибов. Интенсивность проявления сосудистых звездочек повышается при воздействии солнечных лучей. Зачастую данный дефект сочетается с сухостью кожи, гипертрихозом и изменениями, по внешнему виду напоминающими псориаз.
Проблемы с иммунитетом и дыханием
Защитные силы организма на фоне синдрома Луи-Бар значительно ослабевают. Это происходит за счет снижения производства иммуноглобулинов и Т-лимфоцитов. Данные соединения играют важную роль в поддержании клеточного иммунитета.
На фоне снижения защитных сил организма отмечается частое развитие инфекционных процессов, преимущественно поражающих респираторную систему. Дети страдают от ринитов, синуситов, пневмонии и других заболеваний. Для таких патологий характерно длительное течение, а также устойчивость к антибактериальной терапии.
Новообразования
Отдельное место синдрому Луи-Бар в иммунологии отводится еще и потому, что генетическое расстройство зачастую сопровождается высоким риском развития опухолей. Данные процессы чаще всего диагностируются в лимфоретикулярной системе. У пациентов регистрируются раковые поражения красного костного мозга, с трудом поддающиеся лечению. Усугубляется положение и тем фактом, что детям с синдромом Луи-Бар противопоказано использование лучевой терапии. Распространенным заболеванием при данной патологии является лимфома.
Зрение
Телеангиэктазия отмечается не только на коже, но и в мембране, покрывающей склеру глаза. Этот симптом сочетается с поражениями связочного аппарата данного анализатора. Нарушается процесс координации кривизны хрусталика. Вследствие дефектов у детей развивается косоглазие, может снижаться острота зрения.
Ортопедические отклонения
У большинства малышей с атаксией-телеангиэктазией отмечается деформация стоп, что лишь усугубляет двигательные расстройства, поскольку пациентам тяжело переносить вес тела с одной конечности на другую. В ряде случаев диагностируются и различные искривления позвоночника, при этом ярко выраженные проблемы встречаются редко. В случае синдрома Луи-Бара эти дефекты хорошо поддаются хирургической коррекции.
Диагностика
Подтверждение наличия заболевания начинается с осмотра пациента и сбора анамнеза. Патогномоничным считается сочетание нарушений координации с телеангиэктазией. При этом основу диагностики генетических проблем составляет анализ ДНК пациента, который позволяет выявить аномалию строения хромосомы. С точки зрения иммунологии важным считается осуществление анализов крови, в которых выявляют ряд характерных изменений. Они включают в себя:
- Снижение количества лимфоцитов. Это происходит преимущественно за счет уменьшения выработки Т-клеток.
- Недостаточная концентрация иммуноглобулинов. При синдроме Луи-Бар чаще отмечается низкое содержание фракций IgA и IgE.
- Поскольку у некоторых пациентов заболевание сопровождается также симптоматикой аутоиммунных нарушений, отмечается наличие в крови соответствующих комплексов: аутоантител к иммуноглобулинам и митохондриям.
Визуальные методы, позволяющие сделать фото внутренних органов, также широко применяются. Используется УЗИ, МРТ и рентген. Для комплексной оценки состояния больного потребуется консультация врачей различной специализации, начиная с иммунолога и заканчивая ортопедом.

Лечение и прогноз
Специфической терапии, которая позволила бы побороть синдром Луи-Барра, нет. Поэтому борьба с заболеванием носит симптоматический характер. Лечение направлено в первую очередь на недопущение развития инфекционных поражений, которые становятся распространенной причиной гибели пациентов. К летальному исходу приводят и онкологические процессы, контролировать которые очень сложно. Для коррекции состояния пациентов применяются антибиотики, кортикостероидные препараты, витамины и внутривенные инфузии.
Современные концепции лечения недуга основаны на следующих принципах:
- Для борьбы с неврологическими расстройствами используются препараты леводопы, антагонисты дофамина и антихолинергические средства. Коррекция тремора производится медикаментами типа «Габапентина», а «Флуоксетин» и «Буспирон» используются с целью уменьшения интенсивности речевых нарушений.
- Во многих случаях оправдано назначение парентерального питания. Это особенно актуально у маленьких пациентов в период лечения инфекционных поражений.
- Для предотвращения развития септических процессов и осложнений со стороны респираторной системы используются антибиотики широкого спектра действия. Ряд врачей склоняется к оправданности их профилактического назначения.
- Рентгенологические исследования у пациентов с генетическим дефектом строго ограничены. По возможности рекомендуется применять альтернативные методы, например, магнитно-резонансную томографию или ультразвук.
- Для контроля онкологических процессов в организме больных требуется регулярный скрининг. Он подразумевает как проведение стандартных анализов крови, так и тестов с использованием специфических маркеров, которые позволяют распознать опухолевый очаг в лимфоретикулярной системе.
Прогноз при синдроме Луи-Бар неблагоприятный. Большинство больных погибают в 20–25 лет. При этом в 65–70% случаев причиной смерти становится хроническое поражение легких. Инфекции склонны к переходу в септический процесс.
Отзывы
Марина, 32 года, г. Ростов-на-Дону
Сын родился с синдромом Луи-Бар. Это стало заметно, когда он начал учиться ходить. Все время падал, не держал равновесие. Позднее появилась сосудистая сеточка на лице. Врачи поставили предварительный диагноз уже после осмотра. Он подтвердился результатами анализов. Прогноз при болезни неблагоприятный. Сейчас принимаем витаминно-минеральный комплекс, надеемся на лучшее.
Федор, 29 лет, г. Петропавловск-Камчатский
Долго не могли научить дочку ходить. Делала шаг и падала, дрожала голова. Решили обратиться за помощью к врачу. Сдали анализы крови, сделали УЗИ. Доктор поставил диагноз «синдром Луи-Бар». Это генетическая проблема, лечения которой нет. Будем делать все возможное, чтобы поддерживать здоровье дочки.
Загрузка…prosindrom.ru
Синдром Гийена-Барре — причины, симптомы, диагностика и лечение
Синдром Гийена-Барре (СГБ) — острая воспалительная демиелинизирующая полирадикулоневропатия аутоиммунной этиологии. Характерный признак заболевания — периферические параличи и белково-клеточная диссоциация в ликворе (в большинстве случаев). Диагноз синдрома Гийена-Барре устанавливается при наличии нарастающей слабости и арефлексии в более чем 1 конечности. При этом следует исключить другие неврологические заболевания, сопровождающиеся периферическими парезами: полиомиелит, острый период стволового инсульта, токсические поражения ЦНС и др. Лечение пациентов с синдромом Гийена-Барре проводится в стационаре, т. к. больному может потребоваться ИВЛ.
Общие сведения
Синдром Гийена-Барре (СГБ) — острая демиелинизирующая воспалительная полиневропатия аутоиммунной этиологии. Характерный признак заболевания — периферические параличи и белково-клеточная диссоциация в ликворе (в большинстве случаев). В настоящее время в рамках СГБ выделяют четыре основных клинических варианта:
- классическая форма СГБ — острая воспалительная демиелинизирующая полирадикулоневропатия (до 90% случаев)
- аксональная форма СГБ — острая моторная аксональная невропатия. Характерный признак данной формы СГБ — изолированное поражение двигательных волокон. При острой моторно-сенсорной аксональной невропатии поражаются как двигательные, так и чувствительные волокна (до 15%)
- синдром Миллера-Фишера — форма СГБ, характеризующаяся офтальмоплегией, мозжечовой атаксией и арефлексией при слабовыраженных парезах (до 3%)
Помимо вышеуказанных форм синдрома Гийена-Барре, в последнее время выделяют еще несколько атипичных форм заболевания — острую сенсорную невропатию, острую пандизавтономию, острую краниальную полиневропатию, встречающиеся довольно редко.
Клиническая картина синдрома Гийена-Барре
Первыми проявлениями синдрома Гийена-Барре являются, как правило, мышечная слабость и/или сенсорные расстройства (чувство онемения, парестезии) в нижних конечностях, которые, спустя несколько часов (суток) распространяются на верхние конечности. В некоторых случаях заболевание манифестирует болями в мышцах конечностей и пояснично-крестцовой области. Очень редко первым проявлением становятся поражения ЧН (глазодвигательные расстройства, нарушение фонации и глотания). Степень двигательных нарушений при синдроме Гийена-Барре значительно варьируется — от минимальной мышечной слабости до тетраплегии. Парезы, как правило, симметричные и больше выражены в нижних конечностях.
Типична гипотония и существенное снижение (либо полное отсутствие) сухожильных рефлексов. В 30% случаев развивается дыхательная недостаточность. Расстройства поверхностной чувствительности проявляются в виде легкой или умеренной гипо- или гиперкинезии по полиневритическому типу. Приблизительно у половины пациентов наблюдаются расстройства глубокой чувствительности (иногда вплоть до полной ее утраты). Поражения ЧН, выявляемые у большинства больных, проявляются парезом мимических мышц и бульбарными нарушениями. Из вегетативных нарушений наиболее часто наблюдаются сердечные аритмии, артериальная гипертензия, расстройство потоотделения, нарушение функций ЖКТ и тазовых органов (задержка мочи).
Диагноз синдрома Гийена-Барре
При сборе анамнеза необходимо обратить внимание на наличие провоцирующих факторов, так как более чем в 80% случаев развитию СГБ предшествуют те или иные заболевания и состояния (перенесенные инфекции ЖКТ, верхних дыхательных путей, вакцинация, оперативные вмешательства, интоксикация, опухоль). Неврологическое обследование направлено на выявление и оценку выраженности основных симптомов синдрома Гийена-Барре — чувствительных, двигательных и вегетативных расстройств.
Необходимо проведение общеклинических исследований (общий анализ мочи, общий анализ крови), биохимического анализа крови (газовый состав крови, концентрация электролитов сыворотки), исследования ликвора, серологических исследований (при подозрении на инфекционную этиологию заболевания), а также электромиографию, результаты которой имеют принципиальное значение для подтверждения диагноза и определения формы СГБ. В тяжелых случаях заболевания (быстрое прогрессирование, бульбарные нарушения) следует проводить суточное мониторирование АД, ЭКГ, пульсовую оксиметрию и исследование функции внешнего дыхания (спирометрия, пикфлоуметрия).
Для подтверждения диагноза необходимо наличие прогрессирующей мышечной слабости более чем в одной конечности и отсутствие сухожильных рефлексов (арефлексия). Наличие стойких тазовых нарушений, выраженной стойкой ассиметрии парезов, полиморфноядерных лейкоцитов, а также четкого уровня расстройств чувствительности должны вызывать сомнения в диагнозе «синдром Гийена-Барре». Кроме того, существует ряд признаков, абсолютно исключающих диагноз СГБ, среди них: недавно перенесенная дифтерия, симптомы интоксикации свинцом либо доказательства интоксикации свинцом, наличие исключительно сенсорных нарушений, нарушение обмена порфиринов.
Дифференциальный диагноз
В первую очередь синдром Гийена-Барре невролог дифференцирует от иных заболеваний, которые также проявляются периферическими парезами (полиомиелит), а также других полиневропатий. Полиневропатия при острой перемежающейся порфирии может напоминать синдром Гийена-Барре, но, как правило, сопровождается разнообразной психопатологической симптоматикой (галлюцинации, бред) и выраженными абдоминальными болями. Симптоматика, схожая с признаками СГБ, возможна при обширных инсультах ствола головного мозга с развитием тетрапареза, который в острый период принимает черты периферического. Основные отличия миастении от СГБ — вариабельность симптоматики, отсутствие чувствительных расстройств, характерные изменения сухожильных рефлексов.
Лечение синдрома Гийена-Барре
Все пациенты с диагнозом «синдром Гийена-Барре» подлежат госпитализации в стационар с отделением интенсивной терапии и реанимации. Приблизительно в 30% случаев СГБ в виду развития тяжелой дыхательной недостаточности возникает необходимость в ИВЛ, продолжительность которой определяют индивидуально, ориентируясь на ЖЁЛ, восстановление глотания и кашлевого рефлекса. Отключение от аппарата ИВЛ проводят постепенно с обязательным этапом перемежающейся принудительной вентиляции.
В тяжелых случаях с выраженными парезами особое значение для предупреждения осложнений, связанных с длительной обездвиженностью пациента (инфекции, пролежни, тромбоэмболии легочной артерии), имеет правильный уход. Необходима периодическая (не менее одного раза в 2 часа) смена положения пациента, уход за кожей, контроль над функциями мочевого пузыря и кишечника, пассивная гимнастика, профилактика аспирации. При стойкой брадикардии с угрозой развития асистолии может потребоваться установка временного электрокардиостимулятора.
В качестве специфической терапии синдрома Гийена-Барре, направленной на купирование аутоиммунного процесса, в настоящее время применяют пульс-терапию иммуноглобулинами класса G и плазмаферез. Эффективность каждого из методов сравнительно одинакова, поэтому их одновременное применение считается нецелесообразным. Мембранный плазмаферез значительно уменьшает выраженность парезов и продолжительность ИВЛ. Проводят, как правило, 4-6 сеансов с интервалом в один день. В качестве замещающих сред используют 0,9% раствор натрия хлорида или декстран .
Следует помнить о противопоказаниях к проведению плазмафереза (инфекции, нарушения свертываемости крови, печеночная недостаточность), а также о возможных осложнениях (нарушение электролитного состава, гемолиз, аллергические реакции). Иммуноглобулин класса G, как и плазмаферез, уменьшает продолжительность пребывания на ИВЛ; его вводят внутривенно ежедневно в течение 5 дней в дозе 0,4 г/кг. Возможные побочные эффекты: тошнота, головные и мышечные боли, лихорадка.
Симптоматическая терапия при синдроме Гийена-Барре проводится для коррекции нарушений кислотно-основного и водно-электролитного баланса, коррекции уровня артериального давления, профилактики тромбоза глубоких вен и тромбоэмболии. Оперативное вмешательство может понадобиться для трахеостомии в случае продолжительной ИВЛ (более 10 суток), а также гастростомии при тяжелых и длительных бульбарных нарушениях.
Прогноз при синдроме Гийена-Барре
У большинства пациентов с диагнозом «синдром Гийена-Барре» наблюдается полное функциональное восстановление в течение 6-12 месяцев. Стойкая резидуальная симптоматика сохраняется приблизительно в 7-15% случаев. Частота рецидивов СГБ составляет около 4%, летальность — 5%. Возможные причины смерти — дыхательная недостаточность, пневмония или другие инфекции, тромбоэмболия легочной артерии. Вероятность летального исхода в большой степени зависит от возраста пациента: у детей в возрасте до 15 лет она не превышает 0,7%, в ВТО время как у пациентов старше 65 лет достигает 8%.
Профилактика синдрома Гийена-Барре
Специфических методов профилактики синдрома Гийена-Барре не существует. Однако следует уведомить пациента о запрете на прививки в течение первого года от дебюта заболевания, так как любая прививка способна вызвать рецидив заболевания. Дальнейшая иммунизация разрешена, при этом должна быть обоснована ее необходимость. Кроме того, развившийся в течение 6 месяцев после какой-либо вакцинации синдром Гийена-Барре — сам по себе является противопоказанием к применению данной вакцины в будущем.
www.krasotaimedicina.ru
Клинический случай синдрома Луи-Бар Текст научной статьи по специальности «Психиатрия. Психотерапия»
КЛИНИЧЕСКИЙ СЛУЧАЙ СИНДРОМА ЛУИ-БАР
№3-2015
Г.Б. КАДРЖАНОВА, А.Р. СМАГУЛОВА, Г.А. МУХАМБЕТОВА, К.С. САРБАСОВА
Казахский Национальный медицинский университет имениС. Д. Асфендиярова, кафедра нервных болезней. Университетская клиника «Аксай» г. Алматы, Казахстан
УДК 616.8-056.76
В статье приведен случай собственного клинического наблюдения синдрома Луи-Бар (врожденной атаксии-телеангиэктазии) у мальчика 14 лет. Отмечены особенности течения, клинические признаки и дополнительные методы исследования синдрома Луи-Бар.
Ключевые слова: Синдром Луи-Бар, атаксия, телеангиэктазия.
Синдром Луи-Бар_ впервые описан в 1941 году современным французским врачом D. Louis Bar, позднее — американскими врачами E. Boder, R. P. Sedgwick; синоним — атаксия телеангиэктазия. Это — наследственное заболевание, начинающееся в раннем детском возрасте с неврологической симптоматикой, снижением
иммунологической реактивности организма, относится к группе факоматозов. Атаксия-телеангиэктазия — заболевание с аутосомно-рецессивным типом наследования, характеризующееся нарушением репарации ДНК, тяжелым иммунодефицитом, мозжечковой дегенерацией,
телеангиэктазиями различной локализации,
предрасположенностью к онкологическим заболеваниям, сенситивностью к радиационным воздействиям. Частота возникновения 1:40 000 живорожденных. Ген локализован на хромосоме 11 (q22-23). При обследовании отмечаются мозжечковая атаксия, которая обычно становится заметной для окружающих в период приобретения ребенком навыков ходьбы, дизартрия, глазодвигательные нарушения, хореоатетоз, миоклонии, а также эндокринные нарушения. Характерные кожные изменения (телеангиэктазии) появляются в 3—6 лет. Первичная локализация телеангиэктазии — конъюнктива глаз, с последующим распространением на лицо, шею, небо, ушные раковины, дорсальную поверхность рук [1].
Заболевание начинается в раннем детстве, проявляется в первую очередь мозжечковой атаксией (100%). Отмечаются качание головы и туловища, нарушение походки, интенционный тремор и хореоатетоз (90-100%), характерным изменением глаз являются нарушения движения глазных яблока (80-90%), нистагм 99-100%) и косоглазие [2]. При этой редкой форме факоматоза наблюдаются неврологические симптомы, кожные проявления в виде паукообразного разрастания сосудов (телеангиэктазии), снижение иммунологической реактивности организма. Первые признаки болезни появляются в возрасте одного года — четырех лет. Походка становится неустойчивой, появляется неловкость движений, нарушение плавности речи (скандированная речь). Прогрессирование мозжечковых нарушений постепенно приводит к тому, что больные перестают ходить. Дети с атаксией-телеангиэктазией часто болеют простудными заболеваниями, воспалением придаточных пазух носа, воспалением легких. Они обусловлены снижением защитных иммунологических свойств крови, отсутствием специфических антител на фоне прогрессирования болезни нарастают нарушения интеллекта, расстраиваются внимание, память, снижается способность к абстрации. Дети быстро истощаются. Отмечается резкие изменения настроения [3]. Постепенно развиваются атрофии кожи, поседение волоса уже в школьном возрасте, задержка психического и физического развития. Обычны гипоплазия мозжечка, резче выраженная в его черве, гипоплазия вилочковой железы, дисгаммоглобулинемия, поражение мононуклеарных
макрофагов (ретикулезы, лимфосаркомы и др.). Прогноз плохой. Причиной смерти чаще являются хронические заболевания бронхов и легких, лимфомы, карциномы [4]. Нейрорадиологические исследования выявляют
прогрессирующую атрофию мозжечка, преимущественно червя, вторичную атрофическую дилатацию IV желудочка, постгеморрагические очаги в паренхиме мозга, появление которых связано с кровоизлияниями из эктазированных сосудов. Наличие у ребенка атрофических изменений мозжечка, визуализируемых на КТ, МРТ головного мозга, в сочетании с атаксией и прогрессирующим неврологическим дефицитом требует исключения синдрома атаксии-телеангиэктазии [1]. Собственное наблюдение.
Под нашим наблюдением находился мальчик Х., 2000 года рождения, в клинику он поступил в возрасте 14 лет, верифицирован клинический диагноз «синдром Луи-Бар». При поступлении жалобы матери на шаткость при ходьбе, плохо разговаривает, задержку в психическом развитии, общую слабость, часто болеет простудными заболеваниями, частый влажный кашель. Из анамнеза жизни известно, что мальчик родился от 6 беременности, 6 родов. Беременность протекала без особенностей, родился в срок. Вес при рождении 3200,0, закричал сразу, длина тела 50 см. Самостоятельно мальчик стал ходить с 1,5 лет, развивается с задержкой в речевом развитии. Анамнез заболевания: со слов матери мальчик болен с 5 лет, когда впервые обратили внимание, что ребенок стал хуже ходить. В 6 лет пошел в школу, но учиться не смог, с детьми не общался, со школьной программой не справлялся, школу посещал только в течение 1-го месяца. Затем обучался на дому, учебными навыками ребенок не овладел, не научился читать, писать, буквы не знает, при написании букв отмечалось дрожание правой руки. Со временем постепенно нарастала общая слабость, изменилась походка, стал шататься при ходьбе, часто падать, быстро уставать, меньше разговаривать. В 6 летнем возрасте появилось покраснение глаз, телеангиэктазии. Приступов судорог у мальчика не было. С 7 лет появился интенционный тремор в руках. В ноябре 2008 года установлен диагноз: Атаксия телеангиоэктазия. Синдром Луи-Бар. Мальчик неоднократно находился на стационарном лечении в неврологическом отделении по месту жительства. Наследственность отягощена: в семье 1 девочка умерла в возрасте 13 лет — у девочки с 8 лет прогрессивно ухудшалось состояние, нарастала слабость, появилась шаткость при ходьбе, затем тремор рук. С 8 лет перестала ходить, часто болела простудными заболеваниями. Диагноз у девочки не был установлен, симптомы заболевания были аналогичны, как у данного ребенка. В семье еще 3 девочки здоровы (26 лет, 24 года, 17 лет), мальчик 11 лет — здоров. 1 мальчик умер в 8 месяцев от кишечной инфекции. Брак близкородственный (родители — двоюродные брат и сестра), по национальности дунгане. У родителей мальчика 4 внука
№3-2015
от старших дочерей — все здоровы. Брак у детей неродственный.
Состояние мальчика тяжелое по поражению центральной нервной системы. В контакт мальчик вступает замедленно, формально. Поведение пассивное, настроение депрессивное, обращенную речь в пределах быта понимает. Сознание ясное. Когнитивные функции снижены: внимание неустойчивое, память снижена, мышление торпидное, бедная мимика. Навыками чтения и письма не владеет, математические представления и навыки развиты слабо. Запас знаний, представлений ниже возрастной нормы. Темп
речи замедлен, в активе — короткая фраза из 2-3 слов, словарный запас значительно ограничен,
звукопроизношение нарушено. Фонематический слух развит слабо. Состояние артикуляционного аппарата: ограничена подвижность языка и губ. Окружность головы 49 см (-5 см), микроцефальной формы. Черепно-мозговая иннервация -лицо симметричное, движения глазных яблок в полном объеме, горизонтальный нистагм. У мальчика выражены проявления мозжечковой атаксии: ходит при поддержке, походка шаткая с широко расставленными ногами, мегалография (рисунок 1).
Рисунок 1
В позе Ромберга — статическая атаксия. При выполнении пальце-носовой и пяточно-коленной проб отмечается дисметрия, интенционный тремор; положительные феномены Стюарт-Холмса и асинергия Бабинского с 2-х сторон. Мышечный тонус диффузно снижен. Сухожильные рефлексы с рук и ног не вызываются. Имеются симптомы поражения экстрапирамидной системы в виде гипокинезии,
гипомимии, брадилалии, брадипсихии. Мальчик астенического телосложения, пониженного питания, подкожно-жировой слой истончен. Ребенок часто болеет респираторными инфекциями, отстает в физическом развитии от своего возраста в росте и весе: вес 18 кг, рост 130 см. С 2-х сторон отмечаются в области конъюнктивы телеангиэктазии (рисунки 2,3).
Рисунок 2
Рисунок 3
На коже лица и ушных раковин отмечаются пигментные пятна, веснушки на лице, сухость кожи. Видимые слизистые бледные, язык розовый. Периферические лимфатические узлы мелкие. Пульс — 86 в 1 минуту, частота дыхания— 24 в 1 минуту, артериальное давление — 90/60 мм рт.ст. Над легкими перкуторно легочный звук, аускультативно дыхание жесткое, выслушиваются влажные проводные хрипы. Сердечные тоны умеренно приглушены, ритмичны. Живот при пальпации мягкий, безболезненный. Печень и селезенка не увеличены. Стул и диурез не нарушены. Проведено клиническое и лабораторное обследование: Общий анализ кров от 28.04.2015 г: эритроцита 4,52*1012/л, гемоглобин 146 г/л, цветной показатель 0,96, лейкоциты 6,2*10*109/л, сегментоядерные 50, эозинофилы 5, лимфоциты 10, палочкоядерные 35. СОЭ 10 мм/час. (лимфоцитопения). Общий анализ мочи от 28.04.2015 г.:
количество 50 мл, удельный вес 1030, реакция — кислая, РН 6.0, лейкоциты 2-3 в поле зрения. Психолог: Интеллектуальное развитие ниже возрастной нормы. Логопед: Общее недоразвитие речи II уровень. Мозжечковая дизартрия. Компьютерная томография головного мозга от 29.04.2015 г.: на серии КТ сканов в MPR реконструкциях негрубо расширены экстрацеребральные ликворные пространства. Срединные структуры не смещены. Умеренно расширены боковые желудочки. Очаговых изменений в веществе головного мозга не выявлено. В задней черепной ямке расширены субарахноидальные пространства. В намете мозжечка: ликворные образования. Миндалины мозжечка гипоплазированы. Заключение: энцефалопатия с атрофическими изменениями в мозжечке с наличием арахноидальной кисты в намете мозжечка (рисунок 4).
Са*
№3-2015
Рисунок 4
Рентгенологическое исследование органов грудной клетки от 27.04.2015 г.: на рентгенограмме грудной клетки форма ее изменена: бочкообразная с расширением межреберных промежутков в верхних отделах. Очаговые и инфильтративные тени в легких не определяются. Легочный рисунок деформирован, сгущен в медиальных
долях. Корни легких расширены, бесструктурные. Сердце вертикально расположено «капельное», уменьшено в размерах, синусы свободны. Заключение: картина хронического деформирующего бронхита с эмфиземой в стадии обострения (рисунок 5).
Окулист от 28.04.2015 г.: диски зрительных нервов розовые монотонно окрашены, округлые, границы четкие. Вены нормального калибра, извиты, артерии ссужены, умеренно извиты. На конъюнктиве с 2-х сторон телеангиэктазии в виде паукообразного разрастания сосудов. DS Ои Телеангиэктазии. Ангиопатия сетчатки. Выводы. Особенностью данного клинического случая явилось то, что ребенок родился от близкородственного брака с отягощенной наследственностью. Синдром Луи-Бар является аутосомно-рецессивным заболеванием, т. е. проявляется клинически только при получении
к 5
рецессивного гена сразу от обоих родителей. Синдром Луи-Бар это редкое заболевание. В связи со снижением иммунологической реактивности организма пациенты с синдром Луи-Бар должны проходить лечение и наблюдение не только у невролога, но и у иммунолога, отоларинголога, офтальмолога, пульмонолога, эндокринолога, онколога. Детям с синдромом Луи-Бар необходимо проведение иммунограммы с определением Т-супрессоров. С профилактической целью рождения детей с синдромом Луи-Бар рекомендовано братьям и сестрам мальчика не вступать в родственные браки.
СПИСОК ЛИТЕРАТУРЫ
1 А. С. Петрухин. Детская неврология. — М.: 2012. — Т 2. — 560 с.
2 С. И. Козлова. Е. Семанова, Н. С. Денисова, О. Е. Блинникова. Наследственные синдромы и медико-генетическое консультирование. Справочник. — М.: Медицина, 1987. — 320 с.
3 Л. О Бадалян. Детская неврология. — 3-е изд. — М.: Медицина, 1984, — 576 с.
4 Гусев Е. И., Бурд Г. С., Никифоров А.Неврологические симптомы, синдромы, симптомокоплескы и болезни. — М.: Медицина, 1999. — 880 с.
Г. Б. КАДРЖАНОВА, А. Р. СМАГУЛОВА, Г.А. МУХАМБЕТОВА, К. С. САРБАСОВА
ЛУИ-БАР СИНДРОМЫНЫН, КЛИНИКАЛЫК ЖАF ДАЙЫ
ТYЙiн: Ма;алада Луи-Бар синдромымен (туа пайда болган атаксия-телеангиэктазия)14 жастагы баланыц жеке ба;ылаудагы клиникальщ жагдайы керсетшген. Луи-Бар синдромыныц агымы, клиникалы; белгiлерi мен ;осымша зерттеу эдстершщ ерекшелiктерi берiлген.
ТYЙiндi свздер: Луи-Бар синдромы, атаксия, телеангиэктазия
О^ШЙИ ‘-3»
О^Каз®|1У
№3-2015
G.KADRZHANOVA, A. SMAGULOVA, G. MUCHAMBETOVA, K. SARBASOVA
A CLINICAL CASE OF LOUIS-BAR SYNDROME
Resume: The article describes the case of own clinical observations of Louis-Bar syndrome (congenital ataxia-telangiectasia) the boy is 14 years old. Marked features of the course, clinical signs and additional methods of research of Louis-Bar syndrome. Keywords: Louis-Bar syndrome, ataxia, elangiectasia
БОЛЕЗНЬ АЛЬЦГЕЙМЕРА -СЛЕДСТВИЕ ПОРАЖЕНИЯ СОСУДОВ ГОЛОВНОГО МОЗГА
М.Т. МЕРГЕНБАЕВА, А.А. НУРМУХАНБЕТОВА
Казахский Национальный медицинский университет им.С.Д.Асфендиярова
УДК 616.892.32:616.831-005
Обзор представляет собой анализ материалов, посвященных изучению роли поражения сосудов головного мозга, нейродегенеративного процесса, хронической ишемии головного мозга в развитии болезни Альцгеймера и деменции. Генез мнестико-интеллектуальных расстройств обусловлен не столько первично-дегенеративными, сколько сосудистыми изменениями, особенно на уровне микроциркуляторного русла. Кроме того, нейроиммунный компонент усиливает цереброваскулярную патологию, что приводит к деменции и болезни Альцгеймера.
Ключевые слова: болезнь Альцгеймера, постинсультная деменция, когнитивные расстройство, мозговые антигены, бета-амилоид.
В последнее время проблема деменции выходит на одно из первых мест среди причин необратимой инвалидизации и смертности больных. Наряду с ростом сердечно-сосудистых заболеваний и инсультов, увеличивается и частота встречаемости сосудистой, в частности, постинсультной деменции. В наибольшей степени деменция затрагивает лиц пожилого и старческого возраста. В популяции людей 65-79 лет ее распространенность составляет 10-15%, а в возрасте 80 лет и старше достигает 20%. Почти у двух третей больных, выживших после инсульта, наблюдается выраженное в той или иной степени снижение когнитивной функции. Общая распространенность деменции у больных с инсультом составляет от 20% до 25%, при этом она увеличивается с возрастом и составляет около 14% у больных моложе 75 лет и приблизительно 32% — у больных старше 75 лет [1]. Вероятность развития деменции у больных с инсультом возрастает в 4-9 раз, по сравнению с лицами без инсульта. Повторные сосудистые эпизоды приводят к изменению иммунологического ответа на мозговые антигены головного мозга, что в свою очередь приводит к изменению гуморального ответа и формированию аутоиммунной реакции к антигенам мозга. Однако направленность иммунных реакций при острой ишемии головного мозга и хронической недостаточности мозгового кровообращения до настоящего времени недостаточно ясна [2].
Болезнь Альцгеймера в последнее годы приобретает все большее медико-социальное значение. Это заболевание считается наиболее распространенной причиной деменции в пожилом и старческом возрасте. Затраты общества, связанные с болезнью Альцгеймера, сопоставимы с суммарными затратами на онкологические и кардиологические заболевания. Предполагается, что в настоящее время во всем мире болезнью Альцгеймера страдает от 17 до 25 млн. больных, а к 2050 году их число может увеличиться в 4 раза [3].
В настоящее время существует мнение, что болезнь Альцгеймера гетерогенна по своему происхождению: в одних случаях она носит наследственный характер, в других — возникает спорадический. Это заболевание может быть результатом сочетанного действия различных факторов, приводящих в конечном итоге сходным клиническим и
патоморфологическим изменениям [4].R.Stewart
рассматривая имеющиеся данные, свидетельствующие о связи артериальной гипотензии с болезнью Альцгеймера, подчеркивает, что характер данной связи требует уточнения. В частности, наличие деменции сопровождается снижением общего метаболизма, одним из проявлений которого и является пониженное артериальное давление [5].Повышение уровня гомоцистеина является одним из сосудистых факторов риска, на фоне гиперцистеинемии возрастает риск тромбозов, окклюзирующих поражений сосудов и инсульта. В последнее время было показано, что гиперцистеинемия отмечается как при сосудистой деменции, так и при болезни Альцгеймера, что приводит к активации механизмов воспаления, сопровождающихся нейротоксичностью, амилоидогенезом и
микроваскулярными расстройствами [6]. В 1994 году В.Хачински предложил использовать термин «сосудистые когнитивные расстройства» для обозначения нарушений высших мозговых функций вследствие цереброваскулярной патологии. Сосудистые когнитивные расстройства имеют характерные особенности патогенеза и клиники, а также течения, которые позволяют дифференцировать данный вид нарушений когнитивных функций от когнитивных нарушений нейродегенеративной природы, весьма распространенных в популяции [7].
По данным некоторых исследователей, значительные сосудистые изменения обнаруживаются у 48% пациентов болезнью Альцгеймера и у 33% лиц такого же возраста без ее признаков. По данным I.Skoog (2005), болезнь Альцгеймера обнаруживалась в 77% случаев сосудистой деменции и только в 17% случаев сосудистой деменции выявлялась изолированная сосудистая патология. Он указывает, что сочетание болезни Альцгеймера с цереброваскулярной патологией наблюдается у более, чем у 80% больных деменцией [8]. Связь деменции с сосудистым поражением головного мозга доказывается развитием когнитивных нарушений непосредственно после инсульта, либо после преходящих нарушений мозгового кровообращения. При различных нарушениях гомеостаза реакции иммунной системы обусловливают специфическую защиту организма. Однако в некоторых ситуациях иммунологические реакции способствуют развитию
cyberleninka.ru
Синдром атаксии-телеангиэктазии или синдром Луи-Бар — причины, способы лечения
Атаксия-телеангиэктазия — сложное генетическое нейродегенеративное заболевание, которое может проявиться в раннем детстве. Заболевание характеризуется постепенным нарушение координации произвольных движений (атаксия), развитие красноватых поражений кожи и слизистой оболочки из-за постоянного расширения группы кровеносных сосудов (телеангиэктазии) и нарушениями функционирования иммунной системы (например, клеточного и гуморального иммунодефицита), что приводит к повышенной восприимчивости в верхних и нижних дыхательных путях. У людей с атаксией- телеангиэктазией также повышен риск развития определенных злокачественных новообразований, особенно рак лимфатической системы, кроветворных органов (например, лейкемия) или рак мозга.
Прогрессивная атаксия, как правило, развивается в грудном возрасте и поначалу может характеризоваться аномальным расхождением в движениях головы по отношению к туловищу. По мере прогрессирования заболевания это состояние приводит к невозможности нормально двигаться, а иногда даже и ходить к позднему детству или подростковому возрасту. Атаксия часто сопровождается затруднением в произношении слов вследствие нарушения речевого аппарата, а также нарушением способности координировать движения глаз, в том числе возникновением непроизвольных, быстрых, ритмичных движений глаз при попытке сосредоточиться на определенных предметах.
Кроме того, к 6-7 годам у ребенка может появится расширение мелких сосудов кожи, часто появляющиеся на открытых участках кожи, таких как мост носа, уши и определенные области конечностей, а также слизистые оболочки глаз.
Признаки и симптомы синдрома Луи-Бар
Ранним симптомом атаксии-телеангиэктазии является уменьшением мышечной координация, как правило, когда ребенок начинает ходить. Координация (особенно в области головы и шеи) становится нарушенной, и могут возникнуть непроизвольные сокращения мышц. В большинстве случаев психическое функционирование не затрагивается, и большинство детей в умственных способностях никак не отстают от детей без данного заболевания.
Видимые расширенные кровеносные сосуды обычно начинаются в глазах (глаза выглядят кровяным) в возрасте от трех до шести лет, хотя телеангиэктазия может проявится раньше. Эти пятна могут распространиться на веки, лицо, уши, и возможно, другие участки тела. Быстрое моргание глаз и движения, а также поворот головы могут развиваться постепенно. Иногда могут возникать носовые кровотечения. Аденоиды, миндалины и периферические лимфатические узлы могут развиваться аномально или не развиваться вовсе. Мышечная координация в области головы и шеи может быть постепенно нарушена, вызывая рефлексы кашля и проблемы с глотанием, дыханием.
Задержку роста можно объяснить дефицитом гормона роста. Преждевременное старение происходит примерно у девяноста процентов пораженных людей и характеризуется седыми волосами с сухой, тонкой, морщинистой или обесцвеченной кожей в подростковом возрасте.
Из-за нарушения иммунной системы больные с синдромом атаксии-телеангиэктазии подвергаются риску к хроническим или легочным инфекциям, повторяющимся случаям пневмонии и хронического бронхита.
Примерно у одного из трех пораженных людей развивается рак, как правило, рак определенных злокачественных новообразований, особенно лимфатической системы или лейкемии. Воздействие рентгеновских лучей, увеличивает частоту возможных опухолей.
В некоторых случаях может возникнуть легкая форма сахарного диабета. Сахарный диабет — это заболевание, при котором происходит недостаточная выработка гормона инсулина. Первичные симптомы могут проявиться в виде повышенной жажде и мочеиспускание, потерю веса, отсутствие аппетита и усталость.
Опубликовано: 24.04.2017
Сахарный диабет — эндокринное заболевание при котором в крови человека повышается уровень сахара из-за недостаточности гормон производящих клеток поджелудочной железы по-другому инсулина. Из-за отсутствия инсулина организм не может переработать сахар, поступающий с продуктами питания в организм человека. Таким образом вместо прорабатывания сахара в глюкозу, полезный для организма компонент сахар оказывается в прямом виде в организме,
Причины синдрома атаксии-телеангиэктазии
Атаксия телангиэктазия наследуется как аутосомно-рецессивный тип наследования признаков. Генетические заболевания определяются двумя генами, один из которых получен от отца, а другой-от матери.
Рецессивные генетические нарушения происходят, когда человек наследует один и тот же ген для одного и того же признака от каждого родителя.
Ген заболевание, которое вызывает атаксии-телеангиэктазии, известный как ген 11q2/ATM. Хромосомы несут в себе генетические особенности каждого человека. Пары хромосом человека пронумерованы от 1 до 22, с неравной 23-й парой хромосом X и Y для мужчин и двумя хромосомами X для женщин.
Исследователи определили, что ген ATM затрагивает белок, который играет роль в регулировании деления клеток после повреждения ДНК. (ДНК или дезоксирибонуклеиновая кислота является носителем генетического кода.) Белок, который известен как ATM является ферментом, который обычно реагирует на повреждение ДНК, вызывая накопление белка p53, который предотвращает деление клеток. Тем не менее, у лиц с атаксия-телеангиэктазия, патологические изменения в гене вызывает отсутствие или недостаток белка ATM и задерживается накопление белка p53. В результате клетки с повреждением ДНК продолжают разделяться без соответствующего восстановления их ДНК, вызывая повышенный риск развития рака.
Атаксия — ходьба с неустойчивой походкой, вызванная нарушением мышечной координации. Существует множество форм атаксии. Некоторые атаксии передаются по наследству, некоторые имеют другие причины и иногда атаксия может быть симптомом других расстройств. Чтобы найти информацию о других типах атаксии.
Симптомы следующих расстройств могут быть похожими на атаксию-телеангиэктазию. Сравнения могут быть полезны для диагностики:
- Атаксия Фридрейха — это генетическое, прогрессирующее, неврологическое расстройство движения, которое обычно проявляется до подросткового возраста. Начальные симптомы могут включать неправильную осанку, частые падение ребенка и прогрессирующие трудности ходьбы из-за нарушения способности координировать движения. У пациентов с атаксией Фридрейха могут также развиться аномалии некоторых рефлексов; характерные деформации стопы; несогласованность рук; невнятная речь; и быстрые, непроизвольные движения глаз. Атаксия Фридрейха может также быть связана с кардиомиопатией, болезнью сердечной мышцы, которая может характеризоваться одышкой при нагрузке, болью в груди и нарушениями сердечного ритма (сердечные аритмии). В отдельных случаях также может развиться сахарный диабет, состояние, при котором наблюдается недостаточное выделение гормона инсулина. Атаксия Фридрейха может быть унаследована как аутосомно-рецессивный признак.
- Атаксия Пьера-Мари — нервно-мышечный синдром наследуется как доминантный признак. Также известный как болезнь Пьера Мари или наследственная мозжечковая атаксия. Ранним симптомом является неустойчивость, при походке вниз по лестнице или по неровной земле. Частые падения могут произойти по мере того как развиваются сопутствующие симптомы, такие как тремор, потеря координации и невнятность речи. На более поздних стадиях также может произойти небольшая потеря зрения
- Зуб Шарко-Мари-Тута представляет собой группу нарушений, при которых поражаются моторные и сенсорные периферические нервы, что приводит к мышечной слабости и атрофии, в первую очередь в ногах, а иногда и в руках
Диагностика заболевания Луи-Бар
Диагноз атаксия — телеангиэктазия ставят на основании анамнеза пациента, тщательном клиническом обследовании, выявлении характерных симптомов и специальных тестов, включая анализы крови, магнитно-резонансная томография и кариотипирование.
Анализы крови могут обнаружить повышенный уровень сывороточного Альфа-фетопротеина, который обнаруживают примерно в 85% случаев. Анализы крови могут также показать повышенные энзимы печени. Вовремя МРТ магнитное поле и радиоволны используются для создания поперечных изображений мозга, которые могут показать прогрессирующую мозжечковую атрофию. Кариотипирование является специализированным тестом, который обнаруживает хромосомные аномалии, дети с заболеванием Луи-Бар имеют повышенную частоту таких хромосомных отклонений.
Лечение синдрома атаксии-телеангиэктазии
Детям с синдромом Луи-Бар стоит избегать чрезмерного воздействия солнечных лучей. Терапия витамином Е, в некоторых случаях была успешной для временного облегчения некоторых симптомов, но должна проводится только под наблюдением врача для избежание побочных эффектов, также полезно следить за состоянием ребенка и не допускать случаи с авитаминозом, поскольку при данном заболевании иммунная система играет большую роль, например при защите от инфекционных заболеваний.
Больным с подобным синдромом иногда назначают препарат диазепам он может помочь в некоторых случаях, избавится от невнятности речи и непроизвольных сокращений мышц.
Отличная статья 0
med8.ru