
Ген гоша: Характеристика генотипов пациентов с болезнью Гоше I типа в Российской Федерации
Характеристика генотипов пациентов с болезнью Гоше I типа в Российской Федерации
БГ — болезнь Гоше
ген GBA — ген глюкоцереброзидазы
ГЦБ — глюкоцереброзидаза
ЗФТ — заместительная ферментная терапия
МРТ — магнитно-резонансная томография
СЭ — спленэктомия
ЦНС — центральная нервная система
Болезнь Гоше (БГ) относится к группе лизосомных болезней накопления, характеризующихся патологическим отложением в органах и тканях промежуточных продуктов нормального метаболизма вследствие недостаточной активности лизосомных ферментов. При БГ нарушено ферментативное расщепление гликосфинголипидов в результате наследственного дефицита активности кислой β-глюкозидазы (глюкоцереброзидаза — ГЦБ) [1]. В редких случаях причиной БГ является наследственный дефицит белка — активатора ГЦБ сапозина С [2].
БГ — наиболее частая лизосомная болезнь накопления, встречается с частотой от 1:40 000 до 1:60 000 у представителей всех этнических групп; в популяции евреев ашкенази распространенность заболевания достигает 1:450—1:1000.
Накопление нерасщепленных продуктов метаболизма в цитоплазме макрофагов сопровождается продукцией этими клетками провоспалительных цитокинов, аутокринной стимуляцией моноцитопоэза и увеличением абсолютного количества макрофагов в местах их «физиологического дома» (селезенка, печень, костный мозг, легкие), что проявляется увеличением размеров селезенки и печени, инфильтрацией макрофагами костного мозга, легких и других органов [3, 4].
Основными клиническими проявлениями БГ служат спленомегалия, гепатомегалия, цитопения и поражение костей.
Поражение костно-суставной системы при БГ отличается выраженной гетерогенностью и варьирует от бессимптомной остеопении и колбообразной деформации дистальных отделов бедренных костей (колбы Эрленмейера) до тяжелейшего остеопороза и ишемических (асептических) остеонекрозов с развитием вторичных остеоартрозов и, как следствие, необратимых ортопедических дефектов. Именно поражение костно-суставной системы определяет тяжесть течения БГ и качество жизни пациентов.
Стандартом современной диагностики БГ является биохимическое определение активности ГЦБ в лейкоцитах крови (ферментная диагностика). Диагноз подтверждают при снижении активности фермента менее 30% от нормального значения. Степень снижения активности фермента не коррелирует с тяжестью течения заболевания [1].
Молекулярный анализ для выявления мутаций гена GBA позволяет верифицировать диагноз БГ, однако в настоящее время не является обязательным и используется для дифференциальной диагностики сложных клинических случаев или научного анализа [4].
Лечение пациентов с БГ заключается в назначении пожизненной заместительной ферментной терапии (ЗФТ) рекомбинантной глюкозидазой, дозы которой варьируют в зависимости от тяжести клинических проявлений и стадии лечения. Своевременное назначение ЗФТ позволяет остановить прогрессирование болезни и предотвратить необратимое поражение жизненно важных органов. Показания к назначению ЗФТ базируются исключительно на предшествующем эмпирическом опыте и включают детский возраст, выраженную органомегалию, глубокую цитопению, тяжелое поражение костей. Разработка ранних критериев неблагоприятного течения БГ остается актуальной клинической задачей.
Показания к назначению ЗФТ базируются исключительно на предшествующем эмпирическом опыте и включают детский возраст, выраженную органомегалию, глубокую цитопению, тяжелое поражение костей. Разработка ранних критериев неблагоприятного течения БГ остается актуальной клинической задачей.
Целью настоящего исследования явилась характеристика генотипов и генофенотипических корреляций у пациентов с БГ I типа в Российской Федерации.
Материалы и методы
Обследовали 100 взрослых пациентов с БГ I типа. Пациенты находились на обследовании и лечении в научно-клиническом отделении орфанных заболеваний Гематологического научного центр МЗ РФ в период с 2001 по 2012 г. Группу наблюдения составили 41 мужчина и 59 женщин в возрасте от 18 до 80 лет (средний возраст 31 год).
Диагноз БГ был установлен на основании характерной клинической и морфологической картины заболевания и во всех случаях подтвержден ферментной диагностикой — определением активности кислой β-глюкозидазы в лейкоцитах периферической крови. Ферментная диагностика осуществлялась в лаборатории наследственных болезней обмена веществ Медико-генетического научного центра РАМН (зав. — к.м.н. Е.Ю. Захарова).
Ферментная диагностика осуществлялась в лаборатории наследственных болезней обмена веществ Медико-генетического научного центра РАМН (зав. — к.м.н. Е.Ю. Захарова).
У 37 (37%) больных за 1—30 лет до поступления под наше наблюдение проведена диагностическая спленэктомия (СЭ). Клиническая и лабораторная картина БГ существенно отличается у больных, перенесших и не переносивших СЭ (табл. 1).
Тяжесть поражения костно-суставной системы оценивали по клиническим данным (наличие переломов, артрозов, других ортопедических дефектов) и результатам инструментальных исследований: рентгенографии и магнитно-резонансной томографии (МРТ) бедренных костей с захватом тазобедренных и коленных суставов. При необходимости проводили рентгенографию и МРТ других костей, а также денситометрию (двойную энергетическую рентгеновскую абсорбциометрию). На основании результатов инструментальных исследований определяли степень инфильтрации костей, а также наличие остеонекрозов, асептических некрозов головки или шейки бедренных костей, патологических переломов, вторичных остеоартрозов. На основании совокупности указанных данных больных разделили на 4 группы: с легким, умеренным, тяжелым и сверхтяжелым поражением костей.
На основании совокупности указанных данных больных разделили на 4 группы: с легким, умеренным, тяжелым и сверхтяжелым поражением костей.
Согласно данным, представленным в табл. 1, характерными клиническими проявлениями у больных без СЭ (n=63) были спленомегалия (у 100%), тромбоцитопения (94%) и вовлечение костей (100%), однако тяжелое и сверхтяжелое поражение костей выявили лишь у 8 (12,7%) и 1 (1,6%) больных соответственно.
В то же время у остальных 86% больных имелось легкое или умеренное поражение костно-суставной системы без необратимых ортопедических дефектов.
Больные, перенесшие СЭ (n=37), характеризовались наличием гепатомегалии, преимущественно нормальными показателями гемограммы (см. табл. 1). Вместе с тем тяжелое или сверхтяжелое поражение костей определялось у 46 и 5,4% пациентов соответственно, что согласуется с данными литературы, свидетельствующими о неблагоприятном течении БГ у больных, перенесших СЭ [1].
Скрининговое исследование для выявления 4 наиболее частых мутаций гена GBA (N370S, 84GG, L444P, IVS2+1) осуществляли методом аллель-специфической полимеразной цепной реакции в реальном времени.
Результаты и обсуждение
Повреждения гена GBA включают миссенс-мутации, сплайсинговые мутации, вставки, делеции нуклеотидов, кроссоверы между структурным геном и псевдогеном, конверсии генов (негомологичные рекомбинации) [5]. БГ I типа ассоциируется с миссенс-мутациями гена GBA и частичным дефицитом глюкозидазы. Напротив, инактивирующие точковые мутации, рекомбинантные аллели и внутригенные делеции сопряжены с накоплением продуктов деградации гликосфинголипидов в нервной ткани (нейроны, адвентициальные клетки, микроглия) и развитием нейронопатических типов БГ (II и III).
К настоящему времени описано около 350 различных мутаций гена GBA, из которых 4 (N370S, L444P, 84GG, IVS2+1) встречаются наиболее часто и составляют 90% всех мутантных аллелей гена GBA в популяции евреев ашкенази и около 60% мутантных аллелей у больных других этнических групп [5, 6].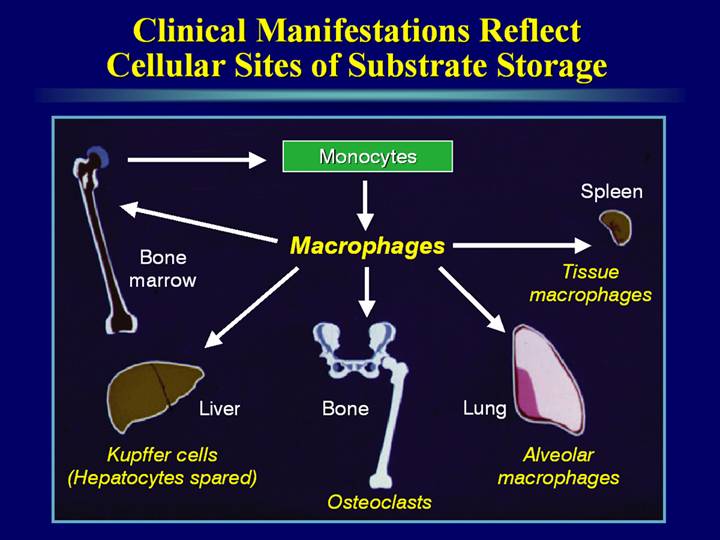
Наиболее распространенная мутация гена GBA при БГ I типа — asn370 → ser (N370S), которая вызвана заменой A на G в позиции 1226 комплементарной ДНК. Эта мутация имеется в 67,2% мутантных аллелей у евреев ашкенази и в 35% у пациентов других национальностей [7]. По данным Международного регистра, наиболее частым генотипом БГ в мире является генотип N370S/N370S [8].
В ходе молекулярно-генетических исследований, проведенных у российских пациентов с БГ, получены следующие результаты. Мутацию N370S выявили у 93 (93%) больных российской группы (Hetero — у 72, Homo — у 21), мутацию L444P hetero — у 21, мутацию IVS2+1 hetero — 2, мутацию 84GG hetero — у 1; в том числе генотип N370S/N370S — у 21, генотип N370S/L444P — у 18, генотип N370S/IVS2+1 — у 2, генотип N370S/? — у 52 пациентов (табл. 2).
Миссенс-мутация leu444 → pro (L444P) вызвана заменой T на С в позиции 1448 комплементарной ДНК. Данная мутация встречается с частотой 3,1% мутантных аллелей в популяции евреев ашкенази и 27,5% аллелей у пациентов нееврейской национальности [7]. По данным Международного регистра БГ, генотип N370S/L444P занимает второе место по распространенности [8].
По данным Международного регистра БГ, генотип N370S/L444P занимает второе место по распространенности [8].
В российской группе мутацию L444P выявили у 21 больного, в том числе генотип N370S/L444P — у 18, L444P/84GG — у 1, L444P/? — у 2.
Мутации, приводящие к нарушению синтеза ГЦБ, в гомозиготном и сочетанном гетерозиготном (84GG/IVS2+1) состоянии являются летальными. Мутация 84GG приводит к сдвигу рамки считывания, что препятствует трансляции глюкозидазы, а мутация IVS2+1 (замена G на А в позиции 1067) нарушает сплайсинг первичного транскрипта вследствие удаления из него 2-го экзона [9].
Мутация 84GG в гетерозиготном состоянии встречается с частотой 12,5% мутантных аллелей в популяции евреев ашкенази и 0,25% — у пациентов других национальностей [7]. В нашей группе гетерозиготную мутацию 84GG выявили у 1 больного.
IVS2+1 в гетерозиготном состоянии встречается с частотой 3,1% мутантных аллелей в популяции евреев ашкенази и практически не встречается среди пациентов нееврейской национальности [7].
В российской группе мутацию IVS2+1 выявили у 2 больных в гетерозиготном состоянии в сочетании с мутацией N370S.
У 4 (4%) больных российской группы ни одной мутации не выявлено.
Результаты предшествующих клинических исследований не выявили корреляций между генотипом и фенотипом БГ [7]. Для выяснения прогностического значения генотипа БГ в российской популяции больных мы провели сравнительный анализ результатов молекулярно-генетических исследований и клинических данных, характеризующих степень тяжести поражения костно-суставной системы (табл. 3). Согласно приведенным данным анализ не выявил очевидной связи между генотипом БГ и тяжестью поражения костно-суставной системы. Генотип N370S/? имелся у больных с легким, умеренным, тяжелым и сверхтяжелым поражением костей примерно с равной частотой. Однако отсутствие информации о втором мутантном аллеле пока не позволяет сделать окончательное заключение.
Заключение
Результаты проведенных клинических и молекулярно-генетических исследований позволили выявить следующее: у 93% российских пациентов с БГ I типа имеется мутация N370S; у 52% больных имеется генотип N370S/?, где второй аллель представлен мутацией, не входящей в число 4 наиболее частых мутаций гена GBA; отсутствует очевидная корреляция между типом мутации гена GBA и тяжестью поражения костно-суставной системы; СЭ приводит к регрессу цитопении, но ассоциируется с последующим тяжелым поражением костно-суставной системы у 51% больных, перенесших СЭ.
В соответствии с этим больным с неясной цитопенией и спленомегалией СЭ следует проводить после исключения диагноза БГ. Пациенты с БГ, подвергавшиеся СЭ, нуждаются в неотложном назначении ЗФТ для предотвращения тяжелого поражения костно-суставной системы и развития необратимых ортопедических дефектов.
Программа диагностики и контроля лечения болезни Гоше (БГ)
В лаборатории наследственных болезней обмена веществ ФГБНУ «Медико-генетический научный центр имени академика Н.П. Бочкова» проводится программа диагностики и контроля лечения болезни Гоше (БГ).
Критерии включения в программу
Общие:
- Гражданство Российской Федерации
- Любой возраст
Наличие более одного из клинических симптомов/изменений лабораторных показателей для болезни Гоше
- Задержка физического развития
- Гепатоспленомегалия, преимущественно спленомегалия
- Тромбоцитопения, анемия
- Спленэктомия в анамнезе
- Носовые кровотечения, экхимозы
В программу принимаются образцы пациентов с установленным ранее диагнозом болезни Гоше для проведения контроля терапии.
Материал для анализа
- Кровь собранная на карточку-фильтр
Подробнее с правилами получения образцов сухой крови на фильтре можно ознакомиться здесь.
Необходимые документы
- Направление на генетическое исследование с указанием предполагаемого диагноза и контактов направившего врача
- подписанное информированное добровольное согласие на медико-генетическое консультирование и генетическое обследование
- подписанное информированное добровольное согласие на обработку персональных данных
- подписанное информированное добровольное согласие на участие в научном исследовании
- подписанное информированное добровольное согласие на обработку персональных данных (научные исследования)
- медицинские документы пациента (краткая выписка с указанием основных клинических симптомов/лабораторных данных, включенных в критерии)
Подробнее о программе
В рамках программы проводится исследование активности бета-глюкоцереброзидазы, а также высокоспецифичных метаболитов характерных для болезни Гоше (lysoGbl1) методом тандемной-масс спектрометрии. Генетическая (поиск мутаций в гене GBA) и дополнительная биохимическая диагностика проводится при выявлении отклонений от нормы по результатам первичной биохимической диагностики.
Генетическая (поиск мутаций в гене GBA) и дополнительная биохимическая диагностика проводится при выявлении отклонений от нормы по результатам первичной биохимической диагностики.
Для вызова курьера, для осуществления транспортировки биологического материала из других городов, звоните по телефону:
Телефон +7 (800) 511 87 66, звонок по России бесплатный
Время работы горячей линии с 10:00 до 19:00 по московскому времени.
Горячая линия предназначена исключительно для медицинских работников.
Также биоматериал и документы можно доставить самостоятельно по адресу: г. Москва, ул. Москворечье д.1, кабинет 235 (регистратура ФГБНУ «МГНЦ»)
Забор биоматериала (услуга платная) для исследования может быть произведен непосредственно в Медико-генетическом научном центре имени академика Н.П. Бочкова по адресу: г. Москва, ул. Москворечье, д. 1, при наличии вышеупомянутых документов, по предварительной записи (запись по телефону +7 (495) 111-03-03.
Дополнительная информация
Сроки проведения биохимической диагностики – 14 рабочих дней, ДНК-диагностики от 25-45 рабочих дней.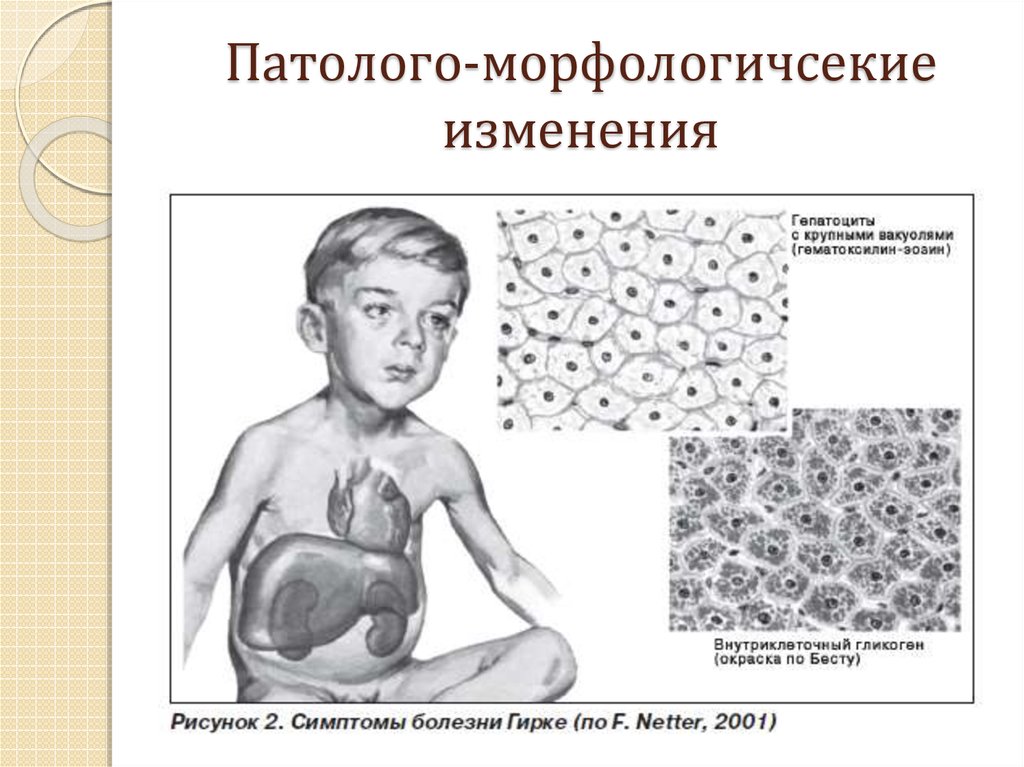
Результаты анализа будут отправлены на адрес/адреса электронной почты, указанные в информированном согласии. ФГБНУ «МГНЦ» не может отправить заключения по запросу врача или представителя пациента, если пациент или его законный представитель не указал в информированном согласие адрес электронной почты.
Если у вас возникнут вопросы по исследованиям вы можете задать их по телефону горячей линии программы +7 (800) 511 87 66
В ФГБНУ «МГНЦ» с 10:00 до 19:00 по московскому времени в рабочие дни работает горячая линия для врачей 8 800 201 09 39.
Успех генной терапии | Больница на Грейт-Ормонд-стрит
Десять лет назад детям с тяжелой и смертельной формой иммунного расстройства в больнице Грейт-Ормонд-Стрит (GOSH) была предоставлена возможность вылечиться с помощью высоко экспериментального и революционного нового лечения — генной терапии. Теперь исследователи из GOSH объявили о результаты многолетней кропотливой работы по обеспечению безопасности этой новаторской техники.
Тяжелый комбинированный иммунодефицит
У детей с нарушениями иммунитета снижена способность бороться с инфекцией. Тяжелый комбинированный иммунодефицит (ТКИН) — ранее смертельная патология — означает, что дети рождаются практически без функционирующей собственной иммунной системы.
Из-за дефекта в одном из нескольких генов, имеющих решающее значение для выработки организмом иммунных клеток, дети с ТКИД не могут атаковать болезнь, а это означает, что любая инфекция, которой они подвергаются, может оказаться фатальной.
Таким образом, эти дети должны быть полностью изолированы от своего окружения в стерильной среде, отсюда и общее название состояния — синдром «мальчика в пузыре».
Генная терапия
«Эти дети были одними из самых больных, которых мы когда-либо видели», — говорит Бобби Гаспар, профессор детской иммунологии в Институте детского здоровья Калифорнийского университета в Лос-Анджелесе и один из членов команды, которая помогла реализовать новаторскую программу генной терапии в больнице Грейт-Ормонд-Стрит. .
.
«Мы знали, что наше лучшее лечение в то время — трансплантация костного мозга — просто не сработает в значительном числе таких случаев, особенно у детей, для которых мы не смогли найти подходящего донора костного мозга».
Решение заключалось в методе, который еще десять лет назад ни разу не испытывался в педиатрии. Он основывался на извлечении образца костного мозга пациента — строительного блока всех иммунных клеток организма — и воздействии на него вирусами, тщательно модифицированными, чтобы нести функционирующую копию неисправного гена, вызывающего SCID.
Затем этот модифицированный вирус инкубируют с костным мозгом, где он переносит и интегрирует функционирующую копию гена в ДНК клеток-реципиентов.
Теперь, вооружившись жизненно важными генетическими инструкциями, которые они ранее упустили, клетки костного мозга повторно вводят пациенту, где они растут, чтобы произвести полный спектр иммунных клеток, необходимых для борьбы с болезнью.
Приверженность инновациям
GOSH проводит больше испытаний генной терапии иммунодефицита у детей, чем любой другой центр в мире.
При постоянной поддержке исследовательская группа надеется развернуть программу генной терапии больницы для ряда других заболеваний, что позволит гораздо большему количеству пациентов предлагать эту новаторскую новую терапию в качестве передового подхода.
История Джека
Джек в своей футбольной форме
Джек Крик, семи лет, был седьмым пациентом новаторской программы X-SCID. Его мама, Дебра, рассказывает его историю.
«Джек родился в мае 2004 года, и в сентябре того же года ему поставили диагноз X-SCID. Он был сопливым, как новорожденный, что врачи объяснили скоплением слизи.
«После того, как нам поставили диагноз, нас госпитализировали в GOSH. При переводе из Лестера здоровье Джека ухудшилось, и когда мы приехали, мы неделю провели в отделении интенсивной терапии.
«После того, как состояние Джека улучшилось, нам сказали, что ему потребуется трансплантация костного мозга (ТКМ), но совпадения найти не удалось. Именно тогда команда обсудила с нами вариант генной терапии.
«К тому моменту у нас действительно не было выбора, потому что у Джека закончились доступные варианты лечения.
«Оглядываясь назад, мы оглядываемся назад и видим, что генная терапия на самом деле была намного эффективнее для Джека, потому что ему не нужна была химиотерапия, которая потребовалась бы при ТКМ.
«Врачи и остальная команда в GOSH замечательные; мы не можем отблагодарить их за их работу и исследования».
Работа по генной терапии финансируется Детской благотворительной организацией больницы Грейт-Ормонд-Стрит, Wellcome Trust, Ассоциацией первичного иммунодефицита и Центром биомедицинских исследований Национального института медицинских исследований в больнице Грейт-Ормонд-Стрит.
GOSH предлагает первое в мире лечение лейкемии | Базовое редактирование и CAR Т-клеточная терапия | Больница на Грейт-Ормонд-стрит
11 декабря 2022 г., 8:00
В мае 2022 года 13-летняя Алисса из Лестера стала первым зарегистрированным пациентом в мире, которому в Детской больнице на Грейт-Ормонд-стрит (GOSH) в сотрудничестве с Детским институтом на Грейт-Ормонд-стрит в Калифорнийском университете была проведена обработка Т-клеток с отредактированной базой. Health (UCL GOS ICH), для лечения ее «неизлечимого» Т-клеточного лейкоза.
Health (UCL GOS ICH), для лечения ее «неизлечимого» Т-клеточного лейкоза.
В 2021 году у Алиссы был диагностирован Т-клеточный острый лимфобластный лейкоз (Т-ОЛЛ), и она получила все современные методы лечения рака, включая химиотерапию и трансплантацию костного мозга. К сожалению, ее болезнь вернулась, и не было никаких других вариантов лечения в рамках обычного ухода.
Алисса была первым пациентом, включенным в новое клиническое исследование, и в мае она была госпитализирована в отделение трансплантации костного мозга (ТКМ) в GOSH для получения генетически модифицированных Т-клеток CAR, которые первоначально были получены от здорового донора. Эти клетки были отредактированы с использованием новой технологии редактирования базы, чтобы позволить им выслеживать и убивать раковые Т-клетки, не атакуя друг друга.
Всего через 28 дней у нее наступила ремиссия, и ей сделали вторую пересадку костного мозга, чтобы восстановить ее иммунную систему. Сейчас, спустя шесть месяцев после ТКМ, она чувствует себя хорошо дома, восстанавливается вместе со своей семьей и продолжает наблюдение после ТКМ в GOSH.
Базовое редактирование и CAR Т-клеточная терапия
Анимационный поясняющий видеоролик о редактировании базы. Иллюстрировано доктором Мариам Кларк (в рамках стажировки NIHR GOSH BRC по исследовательским коммуникациям), под редакцией Мэйси Харди.
Было сложнее лечить Т-клеточный лейкоз с помощью традиционной терапии CAR Т-клетками, потому что Т-клетки, предназначенные для распознавания и атаки раковых Т-клеток, также в конечном итоге убивают друг друга в процессе производства, прежде чем их можно будет использовать в качестве лечения. .
Команды GOSH и UCL GOS ICH работали, используя новую технику редактирования генома, называемую базовым редактированием, над созданием нового типа терапии CAR Т-клеток, способной атаковать раковые Т-клетки. Базовое редактирование работает путем химического преобразования отдельных букв кода ДНК (одиночных нуклеотидных оснований) для изменения Т-клеток.
Команда использовала этот метод для внесения множественных изменений в Т-клетки здорового донора, организованных реестром Энтони Нолана, что означает, что клетки не нужно собирать у пациента:
- что они не подвергаются атаке со стороны собственной иммунной системы пациента
- Удаление «флажка» на модифицированных Т-клетках, что означает, что они не будут атаковать друг друга до того, как их можно будет использовать в качестве лечения
- Удаление второй «флаг», который означает, что клетки невидимы для других методов лечения рака
- Добавление способа для модифицированных клеток распознавать и атаковать раковые Т-клетки
Результатом являются отредактированные Т-клетки CAR, которые можно дать пациенту, чтобы они быстро находили и уничтожали Т-клетки в организма, включая лейкемические Т-клетки. В случае успеха пациент получает трансплантацию костного мозга, чтобы восстановить ослабленную иммунную систему.
Это отличная демонстрация того, как с помощью групп экспертов и инфраструктуры мы можем связать передовые технологии в лаборатории с реальными результатами в больнице для пациентов. На сегодняшний день это наша самая сложная клеточная инженерия, которая прокладывает путь к другим новым методам лечения и, в конечном счете, к лучшему будущему для больных детей. У нас в GOSH и UCL GOS ICH уникальная и особая среда, которая позволяет нам быстро внедрять новые технологии, и мы с нетерпением ждем продолжения наших исследований и предоставления их пациентам, которые в них больше всего нуждаются.
Профессор Васим Касим, профессор клеточной и генной терапии в UCL GOS ICH и консультант-иммунолог в GOSH
История Алисы
С тех пор, как Алисса заболела лейкемией в мае прошлого года, она так и не достигла полной ремиссии.
Доктор Роберт Кьеза, консультант по пересадке костного мозга и терапии CAR Т-клетками в GOSHНи химиотерапией, ни после первой пересадки костного мозга. Только после того, как она получила терапию CD7 CAR-T-клетками и вторую трансплантацию костного мозга в GOSH, она избавилась от лейкемии. Это весьма примечательно, хотя пока это предварительный результат, который необходимо отслеживать и подтверждать в течение следующих нескольких месяцев. Вся команда GOSH очень рада за Алиссу и ее семью, и для меня было честью работать с ними в течение последних нескольких месяцев. Мы были очень впечатлены тем, насколько она смелая, и ничто не делает меня более счастливым, чем видеть ее вне больницы, возвращающейся к более нормальному образу жизни.
Алиссе поставили диагноз Т-клеточный лейкоз в мае 2021 года, после длительного периода, который семья считала простудой, вирусами и общей усталостью.
Пока ее лечили в больницах Лестера и Шеффилда стандартными методами лечения — химиотерапией и трансплантацией костного мозга — к сожалению, врачи не смогли взять ее рак под контроль и добиться ремиссии.
Имея единственный другой вариант паллиативной помощи, Алисса и ее семья подробно обсудили это клиническое испытание с экспертами по BMT и CAR T-клеточной терапии и гематологической службой GOSH и приняли решение первыми попробовать экспериментальное лечение для нее. лейкемия.
Как только я это сделаю, люди так или иначе узнают, что им нужно сделать, поэтому это поможет людям — конечно, я собираюсь это сделать.
Алисса, 13
Алисса — первая пациентка в мире, получившая базовую клеточную терапию, и в настоящее время она находится дома, восстанавливаясь после лечения. Алисса и ее семья уверены, что лейкемия теперь не поддается обнаружению, но знают, что в течение некоторого времени за ней нужно будет внимательно следить.
Киона, мама Алисы сказала :
» Врачи сказали, что первые шесть месяцев самые важные, и мы не хотим быть слишком бесцеремонными, но мы продолжали думать: «Если они могут просто избавиться от этого, просто один раз, она будет в порядке. » И, может быть, мы будем правы ».
“ Алисса очень взрослая, и вы можете забыть, что она всего лишь ребенок, но, надеюсь, это подтвердит исследовательские работы, и они смогут предложить это большему количеству детей — все это должно было быть для чего-то. Это просто кажется таким бессмысленным. Сейчас пытаемся жить между встречами. Алисса хочет вернуться в школу, и скоро это может стать реальностью. Она уже дразнит своего брата, потому что ему пришлось вернуться на осенний семестр. ”
Честно говоря, мы на седьмом небе от счастья — быть дома 9 просто потрясающе.0003 Киона, мама Алиссы
Алисса — первый пациент, получивший это лечение во время испытания, но команда намерена набрать до десяти пациентов с Т-клеточным лейкозом, которые исчерпали все другие варианты лечения. Команды по трансплантации костного мозга (BMT) и терапии CAR T-клетками в GOSH надеются, что, если испытание будет успешным, лечение можно будет предлагать детям на более ранних этапах их лечения. Они также надеются, что этот метод может быть вариантом для других типов лейкемии.
Поддержка исследований
Это исследование финансировалось Советом медицинских исследований. Поддержка более широкой исследовательской программы также исходит от Wellcome, Blood Cancer UK, Национального института исследований в области здравоохранения и ухода (NIHR) и Центра биомедицинских исследований NIHR GOSH.
Уход за Алиссой осуществлялся исследовательской и клинической группой BMT и CAR-T в GOSH при участии врачей, медсестер и смежных специалистов.
Клетки были изготовлены в рамках давней исследовательской программы под руководством профессора Васима Касима из Института детского здоровья UCL Great Ormond Street, который также является почетным консультантом GOSH.
Благодаря раннему финансированию Детской благотворительной организации больницы Грейт-Ормонд-стрит (GOSH Charity) профессор Касим стал пионером в разработке новых методов лечения CAR-Т-клеток с использованием инновационных методов редактирования генов. Дети, партнерство между GOSH и UCL GOS ICH.
Исследовательская группа выражает благодарность Энтони Нолану за предоставление донорских Т-клеток и всех доноров, внесших пожертвования в регистр.
В GOSH мы также разрабатываем планы для нового клинического здания, предназначенного для ухода за детьми и молодыми людьми со всей Великобритании с редкими и трудно поддающимися лечению видами рака. Это будет означать, что наши достижения в исследованиях, уходе и лечении будут предоставляться в специально созданном центре, ориентированном на потребности детей и их семей и позволяющем нам двигаться еще дальше и быстрее в разработке новых и более щадящих методов лечения детей с инвалидностью. рак.
В это исследование принимают только пациентов, имеющих право на лечение NHS. Любые пациенты, имеющие право на лечение в рамках NHS и заинтересованные в этом исследовании, должны обратиться к своему лечащему врачу.